

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Review
Volume 14, Number 5, October 2023, pages 319-333

Subvalvular Aortic Stenosis: Learning From Human and Canine Clinical Research
Amanda E. Croftona ![]() , Samantha L. Kovacsb
, Samantha L. Kovacsb ![]() , Joshua A. Sternc, d
, Joshua A. Sternc, d ![]()
aDepartment of Medicine and Epidemiology, School of Veterinary Medicine, University of California Davis, Davis, CA 95616, USA
bVeterinary Medical Teaching Hospital, School of Veterinary Medicine, University of California Davis, Davis, CA 95616, USA
cDepartment of Clinical Sciences, College of Veterinary Medicine, North Carolina State University, Raleigh, NC 27607, USA
dCorresponding Author: Joshua A. Stern, Department of Clinical Sciences, College of Veterinary Medicine, North Carolina State University, Raleigh, NC 27607, USA
Manuscript submitted July 12, 2023, accepted October 3, 2023, published online October 25, 2023
Short title: SAS: Human and Canine Clinical Research
doi: https://doi.org/10.14740/cr1547
- Abstract
- Introduction and Disease Prevalence
- Dogs as Model Organisms of Human Cardiac Disease
- SAS Through the Centuries
- Pathophysiology and Cardiac Development
- Symptoms, Diagnosis, and Phenotyping
- Clinical Treatment
- Genetics vs. Environment, Mode of Inheritance, and Variant Discovery
- Conclusion and Future Directions
- References
| Abstract | ▴Top |
Subvalvular aortic stenosis (SAS) is the most common congenital heart disease (CHD) in dogs and is also prevalent in human children. A fibrous ridge below the aortic valve narrows the left ventricular outflow tract (LVOT) and increases blood flow velocity, leading to devastating side effects in diseased patients. Due to the similarities in presentation, anatomy, pathophysiology, cardiac development, genomics, and environment between humans and dogs, canine SAS patients represent a critical translational model of human SAS. Potential adverse outcomes of SAS include arrhythmias, left-sided congestive heart failure, endocarditis, exercise intolerance, syncope, and sudden cardiac death. The greatest divergence between canine and human SAS clinical research has been the standard of care regarding treatment of these outcomes, with pharmacological intervention dominating best practices in veterinary medicine and surgical intervention comprising the standard practice for human SAS patients. Regardless of the species, the field has yet to identify a treatment option to prevent disease progression or permanently remove the fibrous ridge, but historical leaps in SAS research support a continued translational approach as the most promising method for achieving this goal.
Keywords: Fixed subaortic stenosis; Discrete subaortic stenosis; Aortic valve; Left ventricular outflow tract; Congenital heart disease; Translational approach; Animal model
| Introduction and Disease Prevalence | ▴Top |
Subvalvular aortic stenosis (SAS), also referred to as subaortic stenosis, is the most common canine congenital heart disease (CHD). SAS represents 24% of canine CHD cases, with 0.3% of canine patients and 4.7% of canine cardiology patients affected in a university teaching hospital setting [1, 2]. This obstructive disease of the left ventricular outflow tract (LVOT) also occurs in human children, making it of translational importance. More specifically, it is diagnosed in 0.25% of children and comprises 6% of human CHD cases [3]. Furthermore, approximately 3-10% of the cases of human CHD are caused by LVOT obstruction, 8-30% of which are cases of discrete subaortic stenosis (DSS); DSS cases also comprise 20% of cases of LVOT obstruction that necessitate intervention [3, 4]. Other species with comparatively high respective prevalences of SAS include pigs and cattle [5].
| Dogs as Model Organisms of Human Cardiac Disease | ▴Top |
Dogs are a naturally occurring model of human CHDs, such as SAS. In comparison to traditional laboratory animals, dogs, as companion animals, occupy environments that are more representative of and consistent with the natural environment of humans [6]. A further benefit to studying large mammalian animal models of heart disease, in comparison to small animal models (such as rodents), is that they have more closely conserved molecular mechanisms to humans [7]. Primates (such as humans) are also phylogenetically more closely related to carnivores (such as dogs) than they are to rodents [8]. For these reasons, dogs provide both an epidemiological and a genetic/genomic advantage as a model of human heart disease. Furthermore, amongst large mammalian animals, the hearts of dogs, in particular, are both anatomically and physiologically similar to human hearts in a number of ways. The three cusps of the aortic valve (left coronary, right coronary, and non-coronary, respectively) have approximately identical dimensions (size, shape, and tissue thickness) in comparison to one another in both humans and dogs, and these dimensions are also comparable between the two species. Additionally, human and canine aortic valves are both anatomically connected to their respective mitral valves via an intervalvar septum (also referred to as a membranous septum), a component of the cardiac fibrous skeleton located between the left fibrous trigone and the central fibrous body [9, 10]. The aortic valves of other large mammalian animal models of heart disease, such as pigs and sheep, do not have the same degree of anatomical similarity to human aortic valves, and sheep do not have an intervalvar (membranous) septum at all. The manner in which our canine companions share many aspects of human anatomy, evolutionarily conserved genes and molecular mechanisms, and even lifestyle makes them an all-around beneficial model organism to aid in our understanding of human cardiac disease and, specifically, of human aortic valve and aortic outflow tract diseases.
This review recounts the history of the collective knowledge built about SAS, and, via a comparative and translational lens, it examines the progress made in both human and veterinary medicine. Specifically, it focuses on our clinical understanding of disease etiology, pathophysiology, presentation, and treatment of SAS in dogs and humans, as well as future directions for areas of ongoing research and innovation.
| SAS Through the Centuries | ▴Top |
The history of aortic valve stenosis is one that spans centuries, as well as species. The pathological process of stenosis, as associated with the aortic valve, was first described in 1663 by Lazare Riviere, a physician who likened the lesions to both carbuncles and hazelnuts and, building upon a Galenic theory of valvular development, suggested that the lesions “hardened by the heat of the heart and changed their substance” [11-13]. In fact, it is credited as the first necropsy-based description of aortic valve pathology [11]. The earliest known record of SAS in the cardiovascular pathology literature was published almost two centuries later, in 1842. Norman Chevers, a physician who, through meticulous postmortem examination of the aortic orifices and valves, noted that the “part of the ‘aortic’ orifice immediately below the valves” is liable to become generally rigid and contracted from inflammatory change,” offered the first of what would become many descriptions (and proposed etiologies) of the development of fibrous tissue and the resulting stenosis below the aortic valve [14]. A mere 2 years later, in 1844, a congenital etiology for aortic stenosis was first proposed, the thought being that a misshapen semilunar valve could be tied to an abnormality in development and, therefore, in the compositional texture of the valve [15, 16].
It was not until the turn of the following century, in 1913, when clinical descriptions of both valvular and myocardial disease were definitively available in the canine cardiovascular disease literature; and until 1925 and 1929, respectively, when more comprehensive pathological details about a portion of recognized canine cardiac diseases had been explored and documented [17, 18]. David Detweiler, widely considered to be the father of veterinary cardiology, was credited by his colleagues as the first veterinarian to promote the study of naturally occurring cardiovascular diseases in dogs as a model for analogous diseases in humans, due to the projected similarities in etiology and pathogenesis across species [18]. In the first half of the 20th century, it was believed that canine cardiovascular diseases, when considered both independently and in aggregate, had a low prevalence in all non-senior/-geriatric life stages and that CHDs in particular were relatively rare [17]. However, no routine methods by which to accurately screen all patients for these diseases had been established, and only certain patients were selected to receive thorough cardiac examinations, so in 1948, Detweiler set out to more definitively characterize canine cardiovascular disease (by defining the categories of disease, as well as their respective prevalences, natural histories, and clinical diagnostic criteria) via diligent clinical and postmortem examinations [17]. As a result of his study, Detweiler and his colleague, Donald Patterson, identified a higher prevalence of cardiovascular disease, distributed more widely across age groups, than had previously been assumed; the preselection criteria for cardiac examination was limiting the observed prevalence of cardiovascular disease and generally revealing only the most severe cases [17]. A mere 3 years later, it was estimated that cardiovascular malformations were diagnosed in approximately 6.8 of every 1,000 patients, per a study at a large university veterinary hospital [6].
Upon further examination, CHDs were observed to be more common in purebred dog breeds, as opposed to mixed breeds. SAS was found to have a higher prevalence in Boxers, German Shepherds, and Newfoundlands, and there were individual families with a particularly high prevalence of the disease, suggesting a genetic etiology, at least in part [6, 17, 19]. Donald Patterson went on to establish the field of veterinary medical genetics [20].
Another giant in her own field, Helen Brooke Taussig, a pioneer in human pediatric cardiology, was one of the first physicians to directly mention canine SAS as a model of human SAS within human medical literature. Her research focus was CHD, and later in her career, after having authored the first CHD-focused textbook, Congenital Malformations of the Heart (1947), she wrote a literature review detailing the clinical research of CHD in the veterinary/animal literature and how it paralleled and could benefit the goals in the clinical research of human CHD [21, 22]. Her hypothesis that CHDs are caused by genetic mutational events that occurred circa the existence of the last common ancestor of all mammals was not shared by many of her contemporaries and has not gained traction. Nevertheless, her conviction that what can be learned by studying the cardiac development of animals and the pathophysiology of naturally occurring CHD in animals can be applied to human medical research ignited further interest in and investigation into canine SAS (among other CHDs), which continues to this day [21, 22]. It furthered the momentum of a translational approach to clinical SAS research.
| Pathophysiology and Cardiac Development | ▴Top |
SAS has two sister diseases, closely related in both a pathological and morphological sense. In all three, an anomalous aggregate of loosely organized fibrous tissue and subendocardial cartilage (deep to the fibrous tissue) forms near or at the location of the aortic valve [5]. In supravalvular aortic stenosis, the fibrous tissue forms craniodorsal to the valve in the canine heart (superior and to the right in the human heart); in valvular aortic stenosis (also referred to as aortic valve stenosis, or simply aortic stenosis), it forms at/on the valve itself; and in SAS (by far the most common of the three in dogs), it typically forms 2 - 5 mm caudoventral to the valve in the canine heart (inferior and to the left in the human heart), frequently protruding 1 - 2 mm into the lumen from the endocardial surface (Fig. 1) [5, 6, 23]. A mild (grade 1) presentation involves one or more nodules (as small as 1 mm) arising from the ventricular septum. In the case of a nodular presentation, nodules are sometimes concurrently appreciated on the ventral aspect of the aortic valve cusps [5, 24]. In a moderate (grade 2) presentation, a ridge/shelf/band forms an arc along a portion of the circumference of the LVOT. The ridge(s) of fibrous tissue tend(s) to extend from the base of the anterior mitral valve leaflet, across the interventricular septum, and below the left coronary cusp of the aortic valve [5, 6, 24]. In severe (grade 3) cases, a ring or “collar” is fully circumscribed in the LVOT [5, 24]. In the most severe cases, one may appreciate a thickening of the ventricular aspects of the aortic valve cusps, as well as the anterior leaflet of the mitral valve [5, 24].
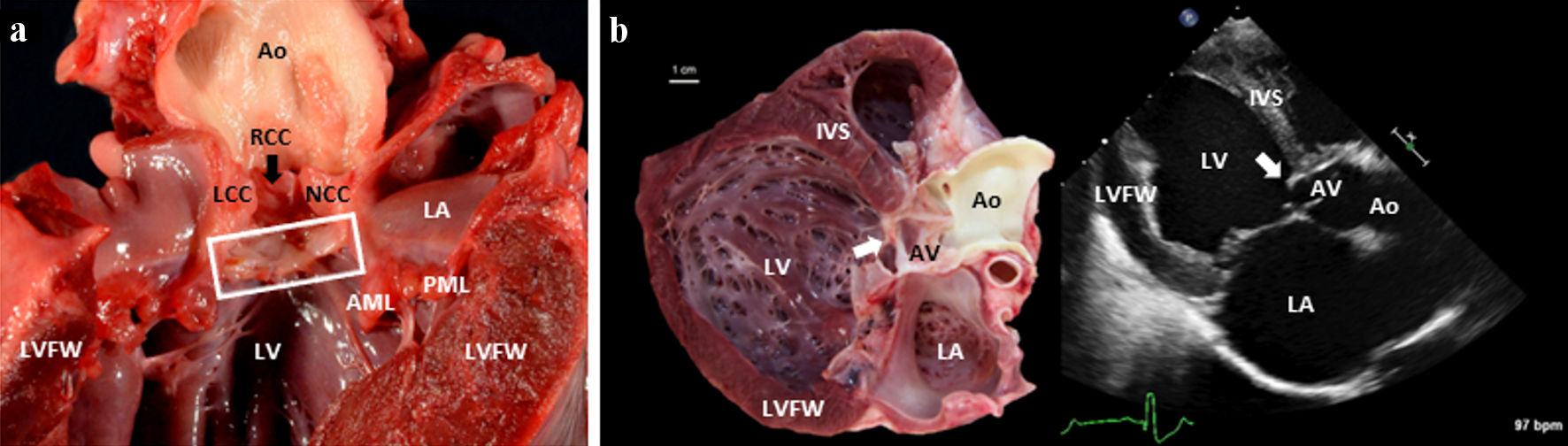 Click for large image | Figure 1. (a) A gross pathology image of the left side of a canine heart affected with SAS. The heart was incised along the long axis of the heart, from the apex to the base (transecting the LVFW). A box surrounds the subvalvular aortic ridge. A black arrow indicates the RCC. (b) A paired gross image and a 2D echocardiographic image (from a right parasternal long-axis five-chamber view) of an 8-month-old Mastiff dog heart afflicted by severe SAS. In both images, you can see the white (gross pathology specimen) and bright (2D echocardiographic image) severe subvalvular ridge/ring of tissue indicated by the white arrow. Note that the specimen shows a dilated and concentrically hypertrophied left ventricle in response to the pressure overload of SAS and the volume overload of concomitant severe AI and mitral valve regurgitation. 2D: two-dimensional; AML: anterior mitral leaflet; Ao: aorta; IVS: interventricular septum; LA: left atrium; LCC: left coronary cusp; LV: left ventricular lumen; LVFW: left ventricular free wall; LVOT: left ventricular outflow tract; NCC: noncoronary cusp; PML: posterior mitral leaflet; RCC: right coronary cusp; SAS: subvalvular aortic stenosis. |
Furthermore, SAS can present as a discrete entity or with a more elongated and diffuse morphology that appears tunnel-like. In terms of location, the lesion could be fused to the ventricular aspect of one or more cusps of the aortic valve or arise either just below the aortic valve or adjacent to the anterior mitral valve leaflet [3, 24]. Whether discrete or tunnel-like in morphology, the obstruction in primary SAS is fixed (also classified as static or resting), meaning that the fibromembranous obstruction is constant and does not vary over time (throughout the course of the cardiac cycle), as it is an anatomical change. This fixed obstruction is the inciting factor for secondary pathophysiological changes, such as left ventricular hypertrophy (LVH). This is in stark contrast to what is sometimes referred to as dynamic (or labile) SAS, in which LVOT obstruction is secondary to a separate primary pathophysiological process and is cardiac cycle phase-dependent. This process could be LVH and resulting protrusion of the left ventricular septum into the LVOT, such as in the case of hypertrophic obstructive cardiomyopathy (HOCM), which is also sometimes referred to as muscular SAS or idiopathic hypertrophic SAS in the human medical literature. Other possibilities are systolic anterior motion (SAM) of the anterior mitral valve leaflet (with or without LVH) or an aortoseptal angle less than 180° [24]. Figure 2 summarizes the classifications of aortic stenosis.
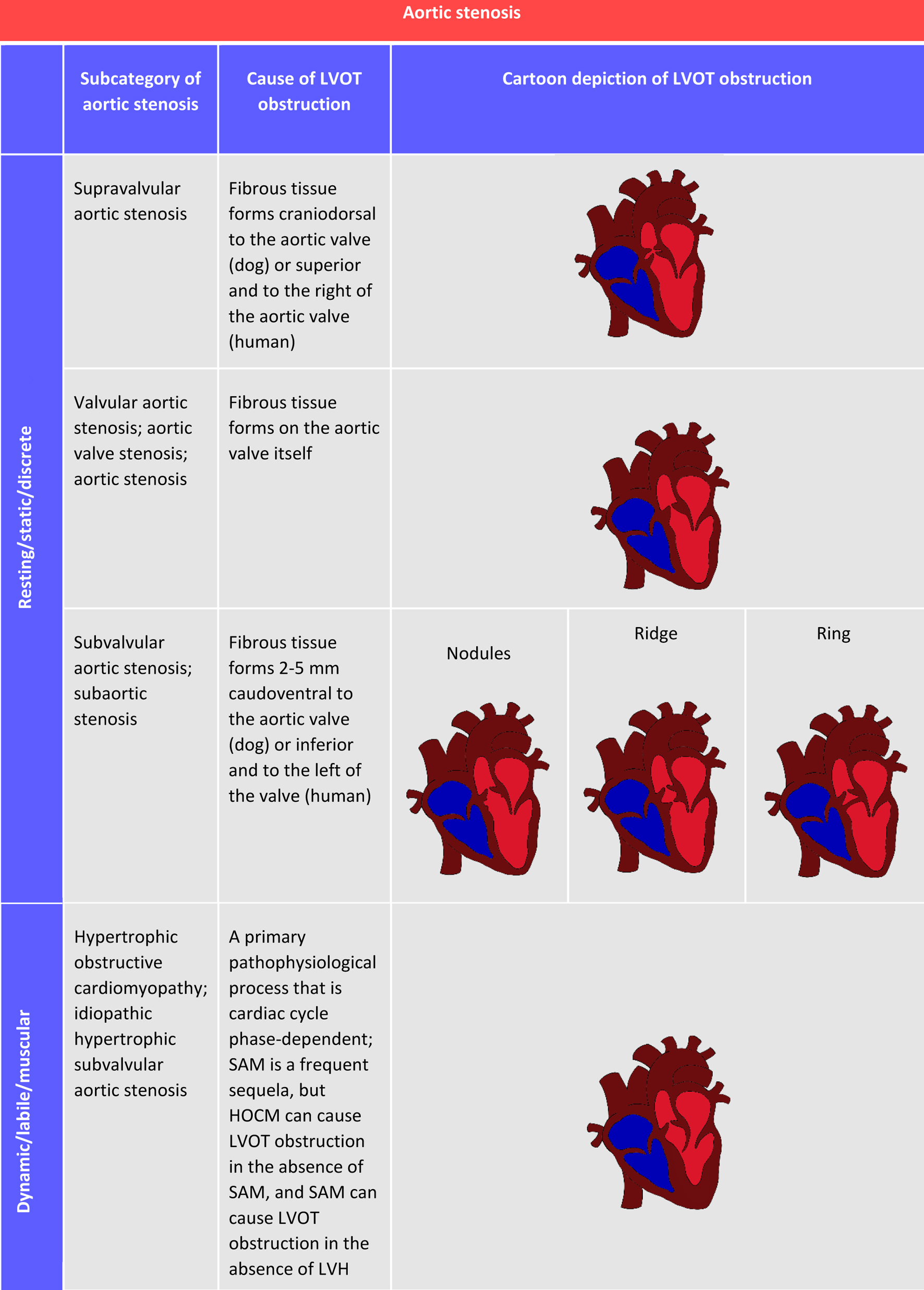 Click for large image | Figure 2. Descriptions and depictions of aortic stenosis in its many forms and the terminology pertinent to this review of subvalvular aortic stenosis. Images are two-dimensional, cross-sectional cartoon illustrations (rotated approximations of right parasternal long-axis five-chamber view). Pathology is illustrated in the context of the human heart. Depictions in a canine heart would be similar, with minor anatomical differences. HOCM: hypertrophic obstructive cardiomyopathy; LVH: left ventricular hypertrophy; LVOT: left ventricular outflow tract; SAM: systolic anterior motion. |
All forms of the disease produce a lesion with five distinct tissue layers, which (starting from the innermost layer that borders the LVOT lumen) are: 1) the endothelium/endocardium; 2) the glycosaminoglycans (GAGs) in the subendothelium; 3) the fibroelastic layer with collagen bundles and elastic fibers; 4) the smooth muscle with a thickened basement membrane; and 5) the fibrous layer with increased amounts of collagen [3]. The numerous collagen fibers that make up the lesion are thickened and oriented irregularly along with short, thin, elastic fibers and occasional fibroblasts [25]. A canine SAS histopathology sample is shown in Figure 3.
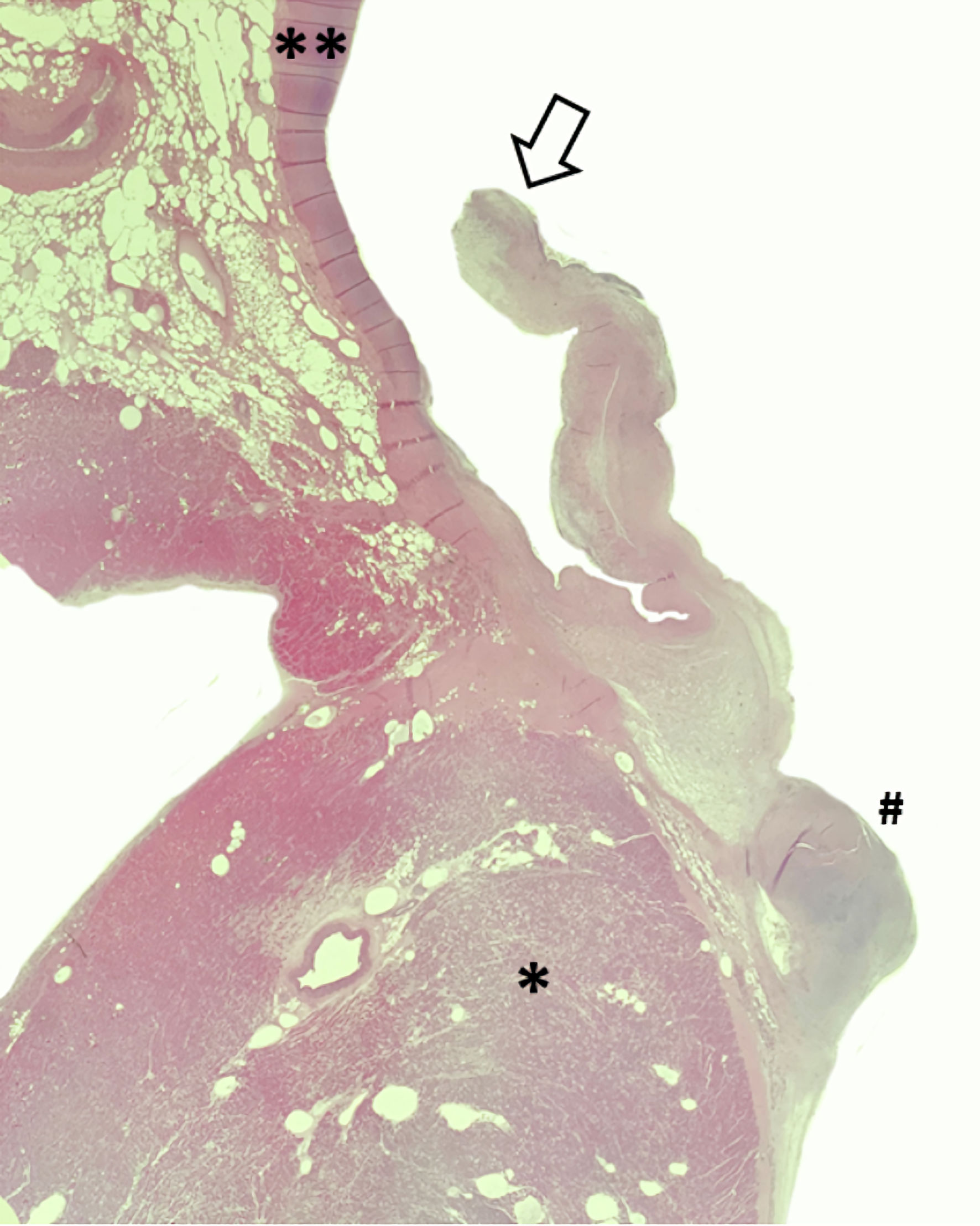 Click for large image | Figure 3. A histopathology sample from a 5-year-old Boerboel with severe SAS stained by hematoxylin and eosin. The open arrow represents an aortic valve leaflet, the double asterisk denotes the aortic wall, the single asterisk is shown within the proximal IVS, and the pound sign denotes the adjacent subvalvular ridge responsible for the increased LVOT pressure gradient. Note the thickening of the aortic valve leaflet in response to turbulent blood flow. IVS: interventricular septum; LVOT: left ventricular outflow tract; SAS: subvalvular aortic stenosis. |
Based upon what is known about the natural history of SAS at this time, it is classified as a congenital disease, as canine patients generally present as young puppies, though debate between a congenital onset and an acquired one remains an active point of debate within the literature. By clinical definition, the disease cannot be diagnosed at birth, as patients cannot be reliably assigned to a severity category until 6 - 12 months of age. While the semilunar valves are formed during embryonic development from the subendocardial tubercles of the truncus arteriosus (at approximately 31 - 35 days in human embryos), the heart continues to undergo developmental changes in the perinatal and neonatal periods [26, 27].
Several congenital comorbidities have been noted in canine SAS patients, including valvular aortic stenosis, supravalvular aortic stenosis, pulmonic stenosis, patent ductus arteriosus, a perimembranous ventricular septal defect, mitral valve stenosis, mitral valve dysplasia, tricuspid valve dysplasia, a bicuspid aorta, a quadricuspid aorta, aortic root hypoplasia, a persistent left cranial vena cava, a double chambered right ventricle, and situs inversus [24, 28]. Likewise, human SAS patients commonly present with a number of concomitant congenital pathological processes, including a bicuspid aortic valve, a ventricular septal defect, and Shone’s complex (a collection of co-occurring conditions that often includes a parachute mitral valve, mitral valve stenosis, a bicuspid aortic valve, and coarctation of the aorta) [3, 29]. This supports the hypothesis that SAS is a congenital disease.
Despite this, the earliest reported cases of canine SAS are diagnosed 3 weeks postnatally, leading some researchers and clinicians to classify the disease as acquired [5]. By contrast, the fact that the disease can, at times, be diagnosed at birth (in human infants) or within later stages of the neonatal period (in puppies and in infants) also serves as support for a congenital onset [18, 30-32]. It should be noted, however, that humans are commonly diagnosed within the first decade of life [33, 34]. A more accurate and specific description might be that SAS is an acquired developmental heart disease with a congenital anatomical substrate, as reported in human medical literature [35]. Pyle et al suggested that the fibrous tissue formed due to the proliferation from and chondrogenic potential of retained embryonic endocardial tissue [5, 24].
More recently, Cape et al detailed the formation of the atypical fibrous tissue that defines SAS and, in doing so, outlined four main components: 1) morphologic abnormalities; 2) altered septal shear stress; 3) genetic predisposition; and 4) cellular proliferation [36]. They proposed a hypothesis in which an anatomical lesion in the region of the LVOT, along with a genetic predisposition, results in a disturbance to laminar flow and a change in septal shear stress. This anatomical lesion could involve an increased distance between the aortic valve and the mitral valve, a reduced (steep) aortoseptal angle, or a diminished LVOT circumference at the level of the aortic annulus [24]. This, in turn, causes changes in the phenotypic expression of mechanosensitive genes of endocardial and endothelial cells and subsequently leads to increased levels of both cell growth and cell division. Simultaneously, the endothelium, injured from altered shear stress, attracts fibroblasts, which then differentiate into myofibroblasts, leading to fibroproliferation and the production of fibrous tissue [35, 36]. Some have postulated that the aortoseptal angle itself or protrusion of the interventricular septum (IVS) into the LVOT represents the inciting lesion of SAS generation across species. In both dogs (particularly noted in the Golden Retriever, Boxer, and Dogue de Bordeaux breeds) and humans, an abnormally steep aortoseptal angle (AoSA), or a malalignment between the aortic root and the IVS, is documented to generate abnormally high shear stress and which could subsequently result in fibrous tissue proliferation [37-41].
As the fibrous tissue emerges below the aortic valve, the diameter of the LVOT at the level of the fibrous tissue decreases. In other words, the shift in the locoregional anatomy results in a narrowing of the LVOT [5, 25, 29, 34, 42-45]. The obstruction to blood flow causes an increase in the aortic blood flow velocity and left ventricular pressure overload ensues [42]. If the pressure is elevated enough, the pressure gradient across the aortic valve may lead to left ventricular concentric hypertrophy, which can subsequently lead to diastolic dysfunction and mitral regurgitation. There may also be subendocardial ischemia/necrosis and fibrosis due to thickened intramural coronary arteries (which may lead to ventricular arrhythmias), left ventricular myocardial failure, and/or left-sided heart failure. It should be noted that LVOT obstruction and coronary arterial thickening may represent separate entities of this disease, as the ischemia and coronary vascular changes of SAS are not proven to be in response to left ventricular (LV) pressure overload alone. In fact, pressure overload models of disease do not directly recapitulate the constellation of coronary vascular change seen across SAS patients of multiple species. Other sequelae of SAS include post-stenotic aortic dilatation and aortic insufficiency (AI) [5].
| Symptoms, Diagnosis, and Phenotyping | ▴Top |
The aforementioned pathology creates a number of antemortem clinical signs and symptoms. Blood flow velocity across the aortic valve increases, resulting in a pressure overload on the left ventricle [42]. Several life-threatening effects can stem from these inciting changes. Potential adverse outcomes of SAS include arrhythmias, left-sided congestive heart failure, endocarditis, exercise intolerance, syncope, and sudden death (due to ventricular arrhythmias from the subendocardial ischemic/fibrotic changes) [5, 46]. In fact, the average lifespan of dogs with severe SAS is a mere 19 months without clinical intervention [47]. Similarly, there is both a high morbidity rate and a high mortality rate among children when their disease is left untreated, but with appropriate intervention, AI is the most prevalent adverse outcome [48].
Phenotyping a patient is performed via a thorough history, a physical examination, and an echocardiogram. In some cases, bloodwork, thoracic radiographs, an electrocardiogram (ECG), and/or a 24-h ambulatory ECG (AECG) may be indicated.
Patients may be asymptomatic, but they may also present with one or more of the following: dyspnea on exertion, angina, effort syncope, presyncope, and orthopnea [25]. The physical examination includes auscultation of the heart for any murmurs or arrhythmias. The typical auscultatory finding in a canine patient is a left basilar systolic heart murmur of ejection character. In a child, the typical auscultatory finding is similar: a low-pitched ejection systolic murmur in the second and third left parasternal spaces, radiating to the suprasternal notch, with an absent ejection click [25]. Other potential findings may include a palpable carotid/left parasternal thrill or a forceful left ventricular apical impulse and a high-pitched early diastolic murmur (if there is AI) [25].
The key feature of an echocardiographic evaluation of dogs with SAS is an assessment of the LVOT and the stenotic region below the aortic valve via two-dimensional (2D), M-mode, and Doppler imaging, as described in published guidelines [43, 49]. This includes right parasternal long-axis views (to screen for a fibrous ridge/ring of fibrous tissue in the LVOT, evaluate heart chamber size and shape, and assess whether there is post-stenotic aortic dilatation), right parasternal short-axis views (to screen for any LVOT narrowing and/or valvular pathology by measuring the diameter at the aortic valve hinge points), and subcostal views (to quantify the maximum LVOT velocity, or aortic velocity, via continuous-wave Doppler measurement) [46]. Left apical continuous wave Doppler imaging is considered an acceptable alternative when subcostal imaging is impaired. Color flow, pulsed wave, and continuous wave Doppler imaging techniques are employed to gauge stenosis severity, to evaluate the presence and magnitude of aortic regurgitation (if any), to assess myocardial function, and to rule out other congenital defects [46]. The severity of a case is defined by the measured aortic velocity and calculated pressure gradient. Although no consensus exists, the authors utilize a generally accepted severity scale where a patient is considered to be SAS-affected with an aortic velocity greater than 2.5 m/s. The severity levels of SAS are further defined by the following aortic velocity cutoffs and pressure gradients, respectively: equivocal (2 - 2.5 m/s; 16 - 24 mm Hg), mild (2.5 - 3.5 m/s; 25 - 49 mm Hg), moderate (3.5 - 4.5 m/s; 50 - 79 mm Hg), and severe (> 4.5 m/s; ≥ 80 mm Hg). Significant variations on the canine echocardiography protocol in human patients include the standard use of three-dimensional (3D) echocardiography and the use of transesophageal echocardiography (TEE) when the patient has a particularly hypertrophied left ventricular septum [25, 50].
Additional methods for assessing severity of SAS and attempting to accurately predict its severity are reported in dogs. One of these methods is the measurement of AoSA. An abnormally steep (low) AoSA, indicating aortoseptal malalignment, has been associated with SAS. One study in Golden Retrievers concluded that while a steep AoSA is not likely to be a reliable single diagnostic for the presence of SAS, it can be a useful contributing factor to the diagnosis and can predict SAS. Some dogs diagnosed with SAS have an abnormally steep AoSA consistent with SAS that is measurable earlier in life than an abnormally high LVOT velocity consistent with SAS [37]. One study in Boxers supports this, concluding that the AoSA of Boxers affected with SAS is, on average, 10° lower than that of Boxers unaffected with SAS [40]. Another of these methods is the measurement of the effective orifice area (EOA), or the area of the cross-section at the narrowest portion of the stenotic jet (the vena contracta). It is approximately 15-20% smaller than the area of the stenotic lesion measured by 2D echocardiographic planimetry and has the advantage of not being flow-dependent, unlike peak and mean pressure gradient measurements. This method aids in predicting disease severity. Unaffected dogs tend to have an indexed EOA (an EOA indexed to body surface area, or BSA) of > 1.25 cm2/m2. Dogs with SAS have significantly smaller EOAs than those of both unaffected adult dogs and unaffected puppies [51, 52].
In human patients, a stenotic aortic valve is also diagnosed echocardiographically and is based on the quantification of the following three hemodynamic parameters: peak jet velocity (determined via continuous-wave Doppler measurement), mean transvalvular pressure gradient, and EOA indexed to BSA [51, 53-57]. When considering the entire stenotic area, the cross-section known as the EOA has the smallest area. Severe aortic stenosis is defined by a peak velocity > 4 m/s, a mean pressure gradient > 40 mm Hg, and/or an EOA < 1 cm2 [53, 54, 57]. ECG results may reveal a prominent Q wave (septal hypertrophy), and radiographs frequently reveal mild cardiomegaly characterized by LVH [25]. Uncommonly, cardiac catheterization is also indicated. It is not utilized to diagnose SAS in and of itself, but the hemodynamic and anatomical evaluation can be particularly helpful in cases where there is thought to be obstruction at multiple levels and as a component of a preoperative workup to rule out and/or characterize concomitant coronary artery disease [25].
| Clinical Treatment | ▴Top |
The treatment of SAS in human and canine patients diverged almost immediately after the identification and recognition of canine SAS within the veterinary medical field - not primarily due to differences in pathology, as one might expect, but, rather, due to differences in anatomy, risks and potential complications, and prioritized treatment outcomes. As a result, humans are generally treated surgically, whereas dogs are generally treated medically.
Aortic balloon valvuloplasty, a minimally invasive interventional option that aims to widen the LVOT via the insertion of an expandable balloon catheter, has been explored in hopes of improving patients’ prognosis. However, in canine SAS patients, the median survival time (MST) with aortic balloon valvuloplasty (55 months) is not significantly different than that with medical management with atenolol (56 months), so this treatment option does not reliably increase patient lifespan [47, 58]. There are reports of cutting balloon valvuloplasty (CBV) combined with high pressure balloon valvuloplasty (HPBV) in the literature, though the long-term clinical benefit, if any, has yet to be determined [59]. Other studies cite the intervention as an option for palliative care [60]. In human patients, percutaneous balloon dilation has been reported occasionally, but results are variable and unreliable, and the procedure is therefore not recommended in these patients. One case report noted a negligible pressure gradient across the aortic valve post-dilation, but it increased sharply after just 12 h [61]. Other possible complications include mitral chord rupture or papillary muscle rupture with secondary mitral regurgitation or pulmonary edema [61]. Balloon valvuloplasty of valvular aortic stenosis in human patients was first reported in 1983 and has since become common practice in the treatment of this related disease [15]. However, neither species responds to balloon valvuloplasty as a method of treatment for SAS, in contrast to other forms of congenital valvular stenosis.
Therefore, the current most commonly employed treatment for SAS in dogs is pharmacological. A beta-adrenergic blocker serves to lower a patient’s heart rate and increase myocardial perfusion; atenolol, which is cardio-selective, tends to be the beta-blocker of choice [42, 62-64]. Treatment is generally selected for dogs with moderate to severe forms of SAS, as mild cases do not often exhibit symptom development or significant lifespan alterations. While balloon valvuloplasty vs. atenolol therapy performed similarly in outcomes assessment for dogs in one clinical trial [58]; a subsequent retrospective study failed to identify a treatment benefit in dogs with SAS receiving atenolol, however, the reliability of retrospective data to answer such a question is questionable [63]. In human medical practice, antihypertensive treatment with beta-blockers is uncommon in patients with severe aortic outflow tract obstructions, as it can commonly result in left ventricular dysfunction and hemodynamic compromise [65]. However, these results are not commonly reported after administration in canine patients, due to physiological differences between humans and dogs, making atenolol a viable treatment option for this species.
A retrospective study has generated some renewed discussion regarding the effectiveness of beta-blockers in these patients, as this study identified no significant survival benefit. However, given its retrospective nature, one could also argue that their lack of difference in survival time between a group of patients with beta-blocker intervention and a control group without the intervention was due to strategic treatment decisions on the part of the treating clinicians (i.e., clinicians may be more prone to withhold or prescribe in the presence of varied disease severity manifestations) [63]. Ultimately, clinicians and researchers have yet to identify a clinical intervention that would alleviate a patient’s stenosis or reverse cardiac remodeling.
However, for human SAS patients, medical treatment alone is not the standard of care. While many patients are started on anti-arrhythmics and cardioprotective therapies, such as beta-blockers, the treatment options carried out in human patients are very different compared to those offered in canine patients. In human patients, the preferred treatment option is surgical repair, involving a resection of the fibrous tissue with the added possibility of a myectomy and a Konno aortoventriculoplasty procedure [25, 34, 66].
Surgical intervention is indicated for approximately two-thirds of human SAS patients [66]. American College of Cardiology/American Heart Association (ACC/AHA) guidelines dictate that surgical intervention is appropriate and indicated for patients with a peak instantaneous echocardiographic gradient greater than 50 mm Hg, a mean gradient greater than 30 mm Hg, or a catheter measurement of the resting peak-to-peak gradient greater than 50 mm Hg [3, 67]. Surgery is further indicated for patients with significant obstruction and for those with a lower pressure gradient secondary to left ventricular systolic dysfunction due to myocardial remodeling [33].
There have been brief ventures into the medical/pharmacological realm to treat human patients with SAS, but only transiently. Ouabain, a cardiac glycoside, was researched in the 1960s as a potential treatment for patients with various forms of aortic stenosis (valvular, subvalvular, and supravalvular) and HOCM. Six patients with aortic stenosis were administered ouabain intravenously via catheterization of the left ventricle. While the increase in myocardial contractility was promising, the lack of significant reduction in end-diastolic pressure made it a poor candidate for the treatment of any form of aortic valve stenosis [68]. The next decade saw the proposition of another medical/pharmacological intervention that was equally short-lived. Propranolol, administered orally to 17 patients with discrete SAS, was found to reduce the intensification of the pressure gradient experienced by SAS patients but was ultimately rejected due to its mere short-acting benefit, eventually overtaken by disease progression [69]. Therein lies the crux of the contrasting methodologies between the treatment of human SAS and that of canine SAS.
Open-heart surgery is seldom performed in dogs, as this intervention is fraught with many challenges irrelevant (or uncommonly relevant) to human SAS intervention and is therefore offered sparsely at a limited number of institutions throughout the world - and, even then, generally only for the treatment of a limited number of diseases. A surgical approach via an aortotomy has been occasionally reported in canine SAS patients, but it has been difficult to simultaneously achieve complete obstructional relief while preventing recurrence, aortic and mitral valve damage, and heart block [70-78]. Additionally, there is poor exposure to the LVOT in dogs via the transaortic approach, and the more fibrocartilaginous nature of the ridge/ring in dogs (compared to the more membranous ridge/ring in humans) is more difficult to resect. An aortotomy also has a high risk of aortic damage [70, 72]. Lastly, and perhaps most importantly, research suggests that the correction of SAS via a transaortic approach does not significantly improve patient lifespan in comparison to the much less invasive pharmacological intervention route, much like aortic balloon valvuloplasty [70, 73, 75]. A modified Konno procedure, including removal of the septal LVOT via a right ventriculotomy, was performed in a case study. The patient’s transaortic pressure gradient decreased from 240 mm Hg preoperatively to 40 mm Hg 2 years postoperatively, at which time the patient was reported to have a good quality of life, as well as LVH regression. However, a larger study that provides data about typical long-term effects is lacking [70].
In recent years, treatment of human aortic stenosis patients with beta-blockers is gaining some level of support, although it is discouraged by the ACC/AHA/American Society of Hypertension (ASH) hypertension guidelines. Specifically, it is contraindicated for patients with chronic AI receive beta-blocker therapy due to the possibility of an increased diastolic filling period, leading to an increase in the severity of valve regurgitation, though this remains a theoretical premise. A retrospective study of 113 patients with severe aortic stenosis revealed that administration of a beta-blocker was associated with a 62% decrease in all-cause mortality [65, 79]. A separate study of 75 patients with moderate to severe AI cited no significant effect of metoprolol, a beta-blocker, on left ventricular volume [65, 80]. Finally, metoprolol has been found to decrease heart rate, aortic valve peak and mean pressure gradients, valvuloarterial impedance, and myocardial oxygen consumption [65]. Therefore, the safety and efficacy of beta-blockers in the treatment of human SAS is being critically re-evaluated.
There is a high recurrence rate after initial surgical relief (estimated to be 6-30% for human patients), despite removal of the anomalous fibrous tissue, suggesting that the underlying pathological process is unaltered [33, 35]. The re-operation rate is also high (23.5%, with 14.7% of patients requiring re-operation due to recurrence, 5.9% due to severe AI, and 2.9% due to an iatrogenic ventricular septal defect (VSD) creation) [34, 66]. Furthermore, the risk of recurrence is greater for human patients who are younger at the time of diagnosis and/or surgical intervention, those who have a smaller aortic annulus, those with an obstruction in close proximity to the aortic valve itself, and those with a higher preoperative peak LVOT pressure gradient [33, 66]. Nevertheless, the majority of the time, survival post-surgery is excellent and not significantly different than people with no diagnosis of SAS [81].
Novel preclinical therapies for SAS are lacking in veterinary patients. Oral rapamycin may represent a promising future direction for the clinical treatment of SAS. Traditionally, rapamycin (sirolimus) has been used as an immunosuppressive agent in organ transplantation [82, 83]. It is an inhibitor of the mammalian target of rapamycin complex 1 (mTORC1) and the mammalian target of rapamycin complex 2 (mTORC2) [83-86]. Growth factor (e.g., IGF-1, or insulin-like growth factor 1) and amino acid ligands act to alter the downstream effects of mTOR on mammalian translational machinery [87-90]. Inhibition of mTORC1, specifically, impedes cell growth (particularly in response to increased workload), proliferation, and differentiation [86]. Rapamycin administration, then, may promote regression of cardiac hypertrophy [87, 91]. Murine research supports a 68% reduction in the heart weight to body weight ratio in mice with compensated hypertrophy and a 41% decrease in the heart weight to body weight ratio with decompensated hypertrophy, when subjects were administered rapamycin under conditions of LV pressure overload. There is also support for enhanced end-diastolic dimensions, fractional shortening, and ejection fraction in mice with decompensated hypertrophy [87]. Rapamycin has also been shown to reduce myocardial infarction and reperfusion injury via cardiac autophagy and anti-oxidative/anti-nitrative stress in mice with cardiac hypertrophy [92]. It remains to be seen if similar clinical outcomes could be achieved in canine and human patients, however. Given these promising responses in conditions of pressure overload and maladaptive hypertrophy, it stands to reason that future investigations of this compound in naturally occurring SAS are warranted. A clinical trial of rapamycin in severe canine SAS is currently underway [93].
Other notable pharmacological treatments that have been explored for an adjacent human disease, aortic stenosis, include verapamil and angiotensin-converting enzyme (ACE) inhibitors. Verapamil is a calcium channel antagonist; it reduces the amount of calcium that enters myocardial cells [94, 95]. It acts as a myocardial depressant and results in more efficient relaxation and diastolic filling in patients with hypertrophic cardiomyopathy (HCM), but these patients with aortic stenosis do not experience these same improvements after verapamil administration. Verapamil has been administered to conscious dogs, but, to the authors’ knowledge, it has not been effectively studied for use in canine aortic stenosis or SAS patients [95]. ACE inhibitors have been contraindicated in patients with aortic stenosis for some time, but evidence for their potential benefit has now surfaced. Angiotensin II is thought to be involved in the development of LVH, myocardial contractile failure, and diastolic dysfunction that make up a trio of sequelae to pressure overload, and in rat models, ACE inhibition appears to dampen these effects [96, 97]. More research is needed to understand the impact for humans with aortic stenosis, as well as human and canine patients with SAS.
Another set of options that could become increasingly critical to the treatment of cardiovascular diseases with known molecular pathways are messenger RNA (mRNA) therapeutics and RNA interference. While the most widely known example of their use is coronavirus disease 2019 (COVID-19) vaccines, mRNA therapeutics have a broad range of applications, including in the treatment of cardiovascular disease. In fact, research teams are currently working on mRNA therapeutics for the treatment of myocardial ischemia, heart failure, arrhythmias, arterial occlusive disease, and hypercholesterolemia. Recent advances, including improvements in mRNA delivery and encapsulation options, have made these therapies more viable [98]. RNA interference (RNAi) is a related but separate treatment option that involves silencing RNA binding to the 3’ untranslated region (UTR) of an mRNA transcript, resulting in decreased levels of translation or increased degradation of that transcript. RNAi treatments for amyloidosis and hypercholesterolemia have been approved; others are in development [98]. The question that we are left with, then, is: Which RNA transcript(s) could be targeted to treat SAS? Part of the key to determining the answer to this question involves answering a related question: Which gene(s) is/are involved in the development of SAS within or across species?
| Genetics vs. Environment, Mode of Inheritance, and Variant Discovery | ▴Top |
The start of canine SAS research did not begin for over a century after the discovery of the disease in humans, but in the 1960s, there was a sudden boom. Clinicians, researchers, and clinician-scientists noted that certain patient populations were disproportionately affected, compared to others. More specifically, it became evident that a few of the larger dog breeds (including German Shepherds, Boxers, and Newfoundlands) had a particularly high prevalence of this disease [6]. Patterson and colleagues concluded that since the distribution of SAS-affected individuals across breeds was non-random, SAS must have a heritable component that affects the development of the heart [6].
Pressing on, Patterson and Pyle performed a series of breeding experiments in Newfoundland dogs, one of the breeds known to have a documented predisposition for SAS. Through this work, they confirmed that the affection status of an individual with regard to SAS is indeed a heritable trait and that the mode of inheritance is not consistent with any Mendelian form of inheritance. Their studies implicate a polygenic mode of inheritance or an autosomal dominant mode of inheritance (possibly with modifiers). These continue to be the leading hypothesized modes of inheritance; additional information is required to more definitively support or refute these contradicting hypotheses [5, 6, 64]. Furthermore, their results suggested that the trait may not have complete specificity, for some crosses between affected individuals yielded offspring with valvular and subvalvular pulmonic stenosis in addition to SAS [5]. A later study would find that Newfoundlands have an odds ratio of 34.73 for the development of SAS, as well as a prevalence of 4.46%. The odds ratio and prevalence, respectively, for other predisposed breeds are as follows: 52.43 and 6.59% for Bullmastiffs, 17.23 and 2.27% for Boxers, 10.67 and 1.42% for Golden Retrievers, 8.78 and 1.17% for Rottweilers, 4.79 and 0.64% for German Shepherds, and 4.59 and 0.61% for Pitbull Terriers [1]. Now, as we have ushered in the 21st century, researchers have begun to elucidate the genetics of SAS in the Bouvier de Flandres, Bullmastiff, Golden Retriever, Newfoundland, and Rottweiler breeds by seeking to identify the genetic variants involved in the molecular pathophysiology of the disease. A three-nucleotide insertion in exon 16 of PICALM (phosphatidylinositol-binding clathrin assembly protein), located on chromosome 11, was discovered in Newfoundlands in 2014 and is the sole variant associated of SAS to date, yet this variant was not replicated in a European breed sample [99, 100]. More recently, a genome-wide association study (GWAS) with the five aforementioned breeds yielded a region of association on chromosome 13 in Golden Retrievers, Newfoundlands, and Rottweilers (both within and across breeds) [64]; chromosome 21 in Bullmastiffs; and chromosome 20 in Bouviers. However, a subsequent whole-genome sequencing (WGS) analysis did not reveal any coding variants within candidate genes to be associated with SAS, as they neither segregated perfectly, nor did their known functions align with the pathophysiological changes established to occur over the course of the disease. Familial occurrence has been observed in humans as well, but specific genetic associations, as well as the mode of inheritance, have yet to be identified [101-107]. Similarly, while the majority of pedigrees in case reports of human families with multiple SAS-affected individuals are consistent with an autosomal dominant or an autosomal recessive mode of inheritance with variable and incomplete penetrance, a conclusive mode of inheritance of SAS in humans is unclear, and the genetic cause of the disease is similarly elusive [105]. Table 1 contrasts these and other research findings and clinical approaches to SAS in human patients with those in canine patients.
 Click to view | Table 1. Comparison Between Canine and Human Disease Characteristics |
Despite this, more extensive knowledge has been garnered from valvular aortic stenosis and supravalvular aortic stenosis. Congenital valvular aortic stenosis and congenital supravalvular aortic stenosis are two diseases that are both anatomically and pathologically similar to congenital SAS and which therefore may provide insight into the disease’s mode of inheritance and/or genomic region(s) associated with SAS. A GWAS revealed three variants associated with valvular aortic stenosis: rs7543130, an intergenic variant on 1p21 near the PALMD (palmdelphin) gene, and rs1830321 (intronic to TEX41, or testis expressed 41, a gene on 2q22 that does not encode a protein); chromatin interaction studies implicated ZEB2 (zinc finger E-box binding homeobox 2) as a potential biological candidate [108]. Meanwhile, a genome-wide DNA methylation analysis found that changes in the methylation patterns at the CpG sites of APOA5 (apolipoprotein A5) and PCSK9 (proprotein convertase subtilisin/kexin type 9) are also associated with valvular aortic stenosis. Conversely, supravalvular aortic stenosis is characterized as an autosomal dominant disease with loss-of-function intragenic mutations or deletions in the ELN (elastin) gene, which result in premature stop codons and/or splicing inefficiency [109-115]. Williams syndrome (or Williams-Beuren syndrome), a complex of conditions that is comprised of supravalvular aortic stenosis and multiple other diseases, is associated with an approximately 1.5 Mb deletion, which includes the 7q11.23 deletion of ELN and contiguous genes in a region of the genome referred to as the WBSCR (Williams-Beuren syndrome critical region) [109-115]. While the aforementioned data serve as a partial blueprint for future SAS-focused genomic explorations, the absence of a definitive genetic etiology of SAS in humans only serves to further highlight the importance of determining the definitive genetic etiology in the canine model of SAS.
| Conclusion and Future Directions | ▴Top |
SAS has been referenced, described, characterized, categorized, reviewed, and analyzed in the medical literature since 1842. Eighteen decades later, through the combined efforts of both veterinary and human medical clinicians and researchers, we have expanded our offerings to our clients and our patients. In the centuries since this disease was first discovered, a vast amount of information has been uncovered. We have much familiarity with its pathology and how it deviates from typical physiology, a mammalian model has been identified in the dog, and we even have the technology to characterize the disease and detail the specifics of a patient’s case noninvasively. Treatment options to address the pathology offer delays in the onset and progression of clinical signs, as well as an increased lifespan, but we have yet to reach the pinnacle of treatment success. The forward momentum built by past researchers leads us to the next frontier of clinical SAS research - identifying a treatment intervention that causes disease progression to either halt or regress. A significant gap in knowledge remains to be bridged - in the clinical realm, the genetic realm, or, more likely, in both.
Acknowledgments
The authors acknowledge Alene Pohly, DVM for providing a photo that illustrates the gross pathology of canine SAS.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Author Contributions
AEC drafted this review article. AEC and JAS edited the manuscript draft. SLK created a portion of the figures. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
2D: two-dimensional; ACC: American College of Cardiology; AECG: ambulatory electrocardiogram; AHA: American Heart Association; AI: aortic insufficiency; AML: anterior mitral leaflet; Ao: aorta; AoSA: aortoseptal angle; APOA5: apolipoprotein A5; ASH: American Society of Hypertension; BSA: body surface area; CBV: cutting balloon valvuloplasty; CHD: congenital heart disease; DSS: discrete subaortic stenosis; ECG: electrocardiogram; ELN: elastin; EOA: effective orifice area; GAG: glycosaminoglycan; GWAS: genome-wide association study; HOCM: hypertrophic obstructive cardiomyopathy; HPBV: high pressure balloon valvuloplasty; IGF-1: insulin-like growth factor 1; IVS: interventricular septum; LA: left atrium; LCC: left coronary cusp; LV: left ventricular; LVFW: left ventricular free wall; LVH: left ventricular hypertrophy; LVOT: left ventricular outflow tract; mRNA: messenger RNA; MST: median survival time; mTORC1: mammalian target of rapamycin complex 1; mTORC2: mammalian target of rapamycin complex 2; NCC: noncoronary cusp; PALMD: palmdelphin; PCSK9: proprotein convertase subtilisin/kexin type 9; PICALM: phosphatidylinositol-binding clathrin assembly protein; PML: posterior mitral leaflet; RCC: right coronary cusp; RNAi: RNA interference; SAM: systolic anterior motion; SAS: subvalvular aortic stenosis; TEE: transesophageal echocardiography; TEX41: testis expressed 41; TXR: thoracic radiograph; WBSCR: Williams-Beuren syndrome critical region; WGS: whole-genome sequencing; ZEB2: zinc finger E-box binding homeobox 2
| References | ▴Top |
- Ontiveros ES, Fousse SL, Crofton AE, Hodge TE, Gunther-Harrington CT, Visser LC, Stern JA. Congenital cardiac outflow tract abnormalities in dogs: prevalence and pattern of inheritance from 2008 to 2017. Front Vet Sci. 2019;6:52.
doi pubmed pmc - Bonagura JD, Twedt DC, Kirk RW. Kirk’s current veterinary therapy XV. Philadelphia, Pennsylvania: Saunders. 2014.
- Masse DD, Shar JA, Brown KN, Keswani SG, Grande-Allen KJ, Sucosky P. Discrete subaortic stenosis: perspective roadmap to a complex disease. Front Cardiovasc Med. 2018;5:122.
doi pubmed pmc - Brink J, Brizard C. Sub-aortic Stenosis. In: Da Cruz EM, Ivy D, Jaggers J, editors. Pediatric and congenital cardiology, cardiac surgery and intensive care [Internet]. London: Springer; 2014. p. 1599-1614.
doi - Pyle RL, Patterson DF, Chacko S. The genetics and pathology of discrete subaortic stenosis in the Newfoundland dog. Am Heart J. 1976;92(3):324-334.
doi pubmed - Patterson DF. Epidemiologic and genetic studies of congenital heart disease in the dog. Circ Res. 1968;23(2):171-202.
doi pubmed - Camacho P, Fan H, Liu Z, He JQ. Large Mammalian Animal Models of Heart Disease. J Cardiovasc Dev Dis. 2016;3(4):30.
doi pubmed pmc - Cannarozzi G, Schneider A, Gonnet G. A phylogenomic study of human, dog, and mouse. PLoS Comput Biol. 2007;3(1):e2.
doi pubmed pmc - Michaelsson M, Ho SY. Congenital heart malformations in mammals: an illustrated text. World Scientific; 2000. p.190.
- Hill AJ, Iaizzo PA. Comparative cardiac anatomy. In: Handbook of Cardiac Anatomy, Physiology, and Devices. Totowa, New Jersey: Humana Press Inc.; 2005.
- Vaslef SN, Roberts WC. Early descriptions of aortic valve stenosis. Am Heart J. 1993;125(5 Pt 1):1465-1474.
doi pubmed - Riviere L. Lazari Riverii, Opera medica universa, quibus continentur. Montpellier, France: Cellier. 1663.
- Sgantzos M, Tsoucalas G, Markatos K, Giatsiou S, Androutsos G. Lazare Riviere (1589-1655 AD), the Pioneer Pharmacologist, Anatomist, and Surgeon, Who Gave the First Modern Description of an Aortic Valve Failure. Surg Innov. 2015;22(5):546-547.
doi pubmed - Chevers N. Observations on the diseases of the orifice and valves of the aorta. Guys Hosp Rep. 1842;7:387-442.
- Hraska V, Photiadis J, Zartner P, Haun C. Congenital aortic valve stenosis and regurgitation. In: Da Cruz EM, Ivy D, Jaggers J, editors. Pediatric and congenital cardiology, cardiac surgery and intensive care [Internet]. London: Springer; 2014. p. 1577-1598.
doi - Paget J. On obstructions of the branches of the pulmonary artery. Med Chir Trans. 1844;27:162-494.164.
doi pubmed pmc - Detweiler DK, Patterson DF. The prevalence and types of cardiovascular disease in dogs. Ann N Y Acad Sci. 1965;127(1):481-516.
doi pubmed - Freedom RM, Yoo SJ, Russell J, Perrin D, Williams WG. Thoughts about fixed subaortic stenosis in man and dog. Cardiol Young. 2005;15(2):186-205.
doi pubmed - Patterson DF. Canine congenital heart disease: epidemiology and etiological hypotheses. J Small Anim Pract. 1971;12(5):263-287.
doi pubmed - Buchanan JW. The history of veterinary cardiology. J Vet Cardiol. 2013;15(1):65-85.
doi pubmed - Gelb BD. History of our understanding of the causes of congenital heart disease. Circ Cardiovasc Genet. 2015;8(3):529-536.
doi pubmed pmc - Taussig HB. World survey of the common cardiac malformations: developmental error or genetic variant? Am J Cardiol. 1982;50(3):544-559.
doi pubmed - Dooley L, Beck C, Bailey S. Mitral Valve Disease. Comparative veterinary anatomy: a clinical approach [Internet]. Academic Press; 2022 [cited Apr 10, 2023]. p. 265-271. Available from: https://reader.elsevier.com/reader/sd/pii/B9780323910156000297?token=73264C716EB3C14D342C319F1AE60E7DCC287057E0A634871010F430160C5962BF5247DF55D72D3E5561A569EA004B63&originRegion=us-east-1&originCreation=20230410103414.
- Petric AD, Perovic A, Svara T, Dovc P. Aortic stenosis in dogs and cats: past, present and future [Internet]. Aortic stenosis - current perspectives. IntechOpen; 2019 [cited Jun 21, 2022]. Available from: https://www.intechopen.com/chapters/undefined/state.item.id.
- Mulla S, Siddiqui WJ. Subaortic Stenosis. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited Sep 30, 2022]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK526085/.
- Kienle RD, Kittleson MD. Chapter 1: Intro. & Cardiac Embryology. In: Small animal cardiovascular medicine. St. Louis, MO: Mosby; 1998.
- Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D, Body SC. Embryonic development of the bicuspid aortic valve. J Cardiovasc Dev Dis. 2015;2(4):248-272.
doi pubmed pmc - Oliveira P, Domenech O, Silva J, Vannini S, Bussadori R, Bussadori C. Retrospective review of congenital heart disease in 976 dogs. J Vet Intern Med. 2011;25(3):477-483.
doi pubmed - Aboulhosn J, Child JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation. 2006;114(22):2412-2422.
doi pubmed - el Habbal MH. Discrete subaortic stenosis in a newborn. Pediatr Cardiol. 1991;12(4):243-244.
doi pubmed - Meyer-Hetling K, Alexi-Meskishvili VV, Dahnert I. Critical subaortic stenosis in a newborn caused by accessory mitral valve tissue. Ann Thorac Surg. 2000;69(6):1934-1937.
doi pubmed - Bilal MS, Oztunc F, Besikci R, Bilal S, Ozkara A, Olga R. Accessory mitral valve tissue causing severe subaortic stenosis with dextrocardia in a premature newborn. Thorac Cardiovasc Surg. 1999;47(4):252-255.
doi pubmed - Kang SH, Kim IJ, Kim WJ. Adult presentation of subaortic stenosis with subaortic membrane treated with surgical removal. J Cardiovasc Dev Dis. 2022;9(2):36.
doi pubmed pmc - Devabhaktuni SR, Chakfeh E, Malik AO, Pengson JA, Rana J, Ahsan CH. Subvalvular aortic stenosis: a review of current literature. Clin Cardiol. 2018;41(1):131-136.
doi pubmed pmc - Papakonstantinou NA, Kanakis MA, Bobos D, Giannopoulos NM. Congenital, acquired, or both? The only two congenitally based, acquired heart diseases. J Card Surg. 2021;36(8):2850-2856.
doi pubmed - Cape EG, Vanauker MD, Sigfusson G, Tacy TA, del Nido PJ. Potential role of mechanical stress in the etiology of pediatric heart disease: septal shear stress in subaortic stenosis. J Am Coll Cardiol. 1997;30(1):247-254.
doi pubmed - Belanger MC, Cote E, Beauchamp G. Association between aortoseptal angle in Golden Retriever puppies and subaortic stenosis in adulthood. J Vet Intern Med. 2014;28(5):1498-1503.
doi pubmed pmc - Buoscio D. Clinical and pathological characterization of unusual form of subvalvular aortic stenosis in four golden retriever puppies. J Am Anim Hosp Assoc. 1994;30:100-110.
- Yap SC, Roos-Hesselink JW, Bogers AJ, Meijboom FJ. Steepened aortoseptal angle may be a risk factor for discrete subaortic stenosis in adults. Int J Cardiol. 2008;126(1):138-139.
doi pubmed - Quintavalla C, Guazzetti S, Mavropoulou A, Bussadori C. Aorto-septal angle in Boxer dogs with subaortic stenosis: an echocardiographic study. Vet J. 2010;185(3):332-337.
doi pubmed - Hollmer M, Willesen JL, Jensen AT, Koch J. Aortic stenosis in the Dogue de Bordeaux. J Small Anim Pract. 2008;49(9):432-437.
doi pubmed - Oyama MA, Thomas WP. Two-dimensional and M-mode echocardiographic predictors of disease severity in dogs with congenital subaortic stenosis. J Am Anim Hosp Assoc. 2002;38(3):209-215.
doi pubmed - Bussadori C, Amberger C, Le Bobinnec G, Lombard CW. Guidelines for the echocardiographic studies of suspected subaortic and pulmonic stenosis. J Vet Cardiol. 2000;2(2):15-22.
doi pubmed - Reist-Marti SB, Dolf G, Leeb T, Kottmann S, Kietzmann S, Butenhoff K, Rieder S. Genetic evidence of subaortic stenosis in the Newfoundland dog. Vet Rec. 2012;170(23):597.
doi pubmed - Vilcant V, Hai O. Left ventricular outflow tract obstruction. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited Apr 10, 2023]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK470446/.
- Kleman ME. Aortic valve stenosis. Veterinary image-guided interventions [Internet]. John Wiley & Sons, Ltd; 2015 [cited Dec 27, 2022]. p. 588-594. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781118910924.ch60.
- Kienle RD, Thomas WP, Pion PD. The natural clinical history of canine congenital subaortic stenosis. J Vet Intern Med. 1994;8(6):423-431.
doi pubmed - Abushaban L, Uthaman B, Selvan JP, Al Qbandi M, Sharma PN, Mariappa TV. Long-term follow-up and outcomes of discrete subaortic stenosis resection in children. Ann Pediatr Cardiol. 2019;12(3):212-219.
doi pubmed pmc - Thomas WP, Gaber CE, Jacobs GJ, Kaplan PM, Lombard CW, Moise NS, Moses BL. Recommendations for standards in transthoracic two-dimensional echocardiography in the dog and cat. Echocardiography Committee of the Specialty of Cardiology, American College of Veterinary Internal Medicine. J Vet Intern Med. 1993;7(4):247-252.
doi pubmed - Oliver JM, Gonzalez A, Gallego P, Sanchez-Recalde A, Benito F, Mesa JM. Discrete subaortic stenosis in adults: increased prevalence and slow rate of progression of the obstruction and aortic regurgitation. J Am Coll Cardiol. 2001;38(3):835-842.
doi pubmed - Belanger MC, Di Fruscia R, Dumesnil JG, Pibarot P. Usefulness of the indexed effective orifice area in the assessment of subaortic stenosis in the dog. J Vet Intern Med. 2001;15(5):430-437.
doi pubmed - DeGroff CG, Shandas R, Valdes-Cruz L. Analysis of the effect of flow rate on the Doppler continuity equation for stenotic orifice area calculations: a numerical study. Circulation. 1998;97(16):1597-1605.
doi pubmed - Adda J, Stanova V, Habib G, Rieu R. In vitro correlation between the effective and geometric orifice area in aortic stenosis. J Cardiol. 2021;77(4):334-340.
doi pubmed - Baumgartner HC, Hung JC-C, Bermejo J, Chambers JB, Edvardsen T, Goldstein S, Lancellotti P, et al. Recommendations on the echocardiographic assessment of aortic valve stenosis: a focused update from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. Eur Heart J Cardiovasc Imaging. 2017;18(3):254-275.
doi pubmed - Garcia D, Pibarot P, Dumesnil JG, Sakr F, Durand LG. Assessment of aortic valve stenosis severity: A new index based on the energy loss concept. Circulation. 2000;101(7):765-771.
doi pubmed - Skjaerpe T, Hegrenaes L, Hatle L. Noninvasive estimation of valve area in patients with aortic stenosis by Doppler ultrasound and two-dimensional echocardiography. Circulation. 1985;72(4):810-818.
doi pubmed - Bonow RO, Carabello B, de Leon AC, Jr., Edmunds LH, Jr., Fedderly BJ, Freed MD, Gaasch WH, et al. Guidelines for the management of patients with valvular heart disease: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients with Valvular Heart Disease). Circulation. 1998;98(18):1949-1984.
doi pubmed - Meurs KM, Lehmkuhl LB, Bonagura JD. Survival times in dogs with severe subvalvular aortic stenosis treated with balloon valvuloplasty or atenolol. J Am Vet Med Assoc. 2005;227(3):420-424.
doi pubmed - Kleman ME, Estrada AH, Maisenbacher HW, 3rd, Prosek R, Pogue B, Shih A, Paolillo JA. How to perform combined cutting balloon and high pressure balloon valvuloplasty for dogs with subaortic stenosis. J Vet Cardiol. 2012;14(2):351-361.
doi pubmed - Sykes KT, Gordon SG, Saunders AB, Vitt JP, O'Brien MT, Fries RC. Palliative combined cutting and high-pressure balloon valvuloplasty in six dogs with severe, symptomatic subaortic stenosis. J Vet Cardiol. 2020;31:36-50.
doi pubmed - Moorthy N, Ananthakrishna R, Rao DPV, Manjunath SC, Nanjappa MC. Percutaneous balloon dilation of discrete subaortic stenosis: a futile exercise. J Invasive Cardiol. 2017;29(9):E107-E108.
pubmed - Galderisi M, D'Errico A. Beta-blockers and coronary flow reserve: the importance of a vasodilatory action. Drugs. 2008;68(5):579-590.
doi pubmed - Eason BD, Fine DM, Leeder D, Stauthammer C, Lamb K, Tobias AH. Influence of beta blockers on survival in dogs with severe subaortic stenosis. J Vet Intern Med. 2014;28(3):857-862.
doi pubmed pmc - Ontiveros ES, Stern JA. Genetics of canine subvalvular aortic stenosis (SAS). Canine Med Genet. 2021;8(1):4.
doi pubmed pmc - Kang TS, Park S. Antihypertensive treatment in severe aortic stenosis. J Cardiovasc Imaging. 2018;26(2):45-53.
doi pubmed pmc - Lopes R, Lourenco P, Goncalves A, Cruz C, Maciel MJ. The natural history of congenital subaortic stenosis. Congenit Heart Dis. 2011;6(5):417-423.
doi pubmed - Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, Del Nido P, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: Executive Summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines for the management of adults with congenital heart disease). Circulation. 2008;118(23):2395-2451.
doi pubmed - Braunwald E, Brockenbrough EC, Frye RL. Studies on digitalis. V. Comparison of the effects of ouabain on left ventricular dynamics in valvular aortic stenosis and hypertrophic subaortic stenosis. Circulation. 1962;26:166-173.
doi pubmed - Adelman AG, Shah PM, Gramiak R, Wigle ED. Long-term propranolol therapy in muscular subaortic stenosis. Br Heart J. 1970;32(6):804-811.
doi pubmed pmc - Nelson DA, Fossum TW, Gordon S, Miller MW, Felger MC, Mertens MM, McMichael M, et al. Surgical correction of subaortic stenosis via right ventriculotomy and septal resection in a dog. J Am Vet Med Assoc. 2004;225(5):705-708.
doi pubmed - Jahangiri M, Nicholson IA, del Nido PJ, Mayer JE, Jonas RA. Surgical management of complex and tunnel-like subaortic stenosis. Eur J Cardiothorac Surg. 2000;17(6):637-642.
doi pubmed - Komtebedde J, Ilkiw JE, Follette DM, Breznock EM, Tobias AH. Resection of subvalvular aortic stenosis. Surgical and perioperative management in seven dogs. Vet Surg. 1993;22(6):419-430.
doi pubmed - Monnet E, Orton EC, Gaynor JS, Boon J, Wagner A, Linn K, Eddleman LA, et al. Open resection for subvalvular aortic stenosis in dogs. J Am Vet Med Assoc. 1996;209(7):1255-1261.
pubmed - Levitt L. Aortic stenosis in the dog: a review of 12 cases. J Am Anim Hosp Assoc. 1989;25:357-362
- Orton EC, Herndon GD, Boon JA, Gaynor JS, Hackett TB, Monnet E. Influence of open surgical correction on intermediate-term outcome in dogs with subvalvular aortic stenosis: 44 cases (1991-1998). J Am Vet Med Assoc. 2000;216(3):364-367.
doi pubmed - Baird DK, Duffell SJ. Resection of a fibromuscular subaortic stenosis in a dog. J Small Anim Pract. 1974;15(1):37-41.
doi pubmed - Muir GD, Panciera DL, Fowler JD, Bharadwaj BB, Burrows P. Medical and surgical management of aortic stenosis in a dog. Can Vet J. 1989;30(11):894-896.
pubmed pmc - Eyster GE, Hough JD, Evans AT, Anderson LK, Carrig CB, Zimmer M, O'Handley P, et al. Surgical repair of patent ductus arteriosus, aortic stenosis, and aortic regurgitation in a dog. J Am Vet Med Assoc. 1975;167(10):942-944.
pubmed - Rossi A, Temporelli PL, Cicoira M, Gaibazzi N, Cioffi G, Nistri S, Magatelli M, et al. Beta-blockers can improve survival in medically-treated patients with severe symptomatic aortic stenosis. Int J Cardiol. 2015;190:15-17.
doi pubmed - Broch K, Urheim S, Lonnebakken MT, Stueflotten W, Massey R, Fossa K, Hopp E, et al. Controlled release metoprolol for aortic regurgitation: a randomised clinical trial. Heart. 2016;102(3):191-197.
doi pubmed - van der Linde D, Roos-Hesselink JW, Rizopoulos D, Heuvelman HJ, Budts W, van Dijk AP, Witsenburg M, et al. Surgical outcome of discrete subaortic stenosis in adults: a multicenter study. Circulation. 2013;127(11):1184-1191.e1181-1184.
doi pubmed - Parlakpinar H, Gunata M. Transplantation and immunosuppression: a review of novel transplant-related immunosuppressant drugs. Immunopharmacol Immunotoxicol. 2021;43(6):651-665.
doi pubmed - Wang L, Gout I, Proud CG. Cross-talk between the ERK and p70 S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in cardiomyocytes. J Biol Chem. 2001;276(35):32670-32677.
doi pubmed - Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, et al. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214(2):316-321.
doi pubmed - Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073-1082.
doi pubmed - Wu X, Cao Y, Nie J, Liu H, Lu S, Hu X, Zhu J, et al. Genetic and pharmacological inhibition of Rheb1-mTORC1 signaling exerts cardioprotection against adverse cardiac remodeling in mice. Am J Pathol. 2013;182(6):2005-2014.
doi pubmed - McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109(24):3050-3055.
doi pubmed - Thomas G, Hall MN. TOR signalling and control of cell growth. Curr Opin Cell Biol. 1997;9(6):782-787.
doi pubmed - Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103(2):253-262.
doi pubmed - Lavandero S, Foncea R, Perez V, Sapag-Hagar M. Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 1998;422(2):193-196.
doi pubmed - Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2(3):e004796.
doi pubmed pmc - Ma LL, Ma X, Kong FJ, Guo JJ, Shi HT, Zhu JB, Zou YZ, et al. Mammalian target of rapamycin inhibition attenuates myocardial ischaemia-reperfusion injury in hypertrophic heart. J Cell Mol Med. 2018;22(3):1708-1719.
doi pubmed pmc - Morris Animal Foundation [Internet]. [cited Feb 21, 2023]. Investigating a novel drug therapy for heart disease. Available from: https://www.morrisanimalfoundation.org/study/heart%20disease-rapamycin-dogs.
- Hess OM, Murakami T, Krayenbuehl HP. Does verapamil improve left ventricular relaxation in patients with myocardial hypertrophy? Circulation. 1986;74(3):530-543.
doi pubmed - Newman RK, Bishop VS, Peterson DF, Leroux EJ, Horwitz LD. Effect of verapamil on left ventricular performance in conscious dogs. J Pharmacol Exp Ther. 1977;201(3):723-730.
pubmed - Goel SS, Kleiman NS, Zoghbi WA, Reardon MJ, Kapadia SR. Renin-angiotensin system blockade in aortic stenosis: implications before and after aortic valve replacement. J Am Heart Assoc. 2020;9(18):e016911.
doi pubmed pmc - Routledge HC, Townend JN. ACE inhibition in aortic stenosis: dangerous medicine or golden opportunity? J Hum Hypertens. 2001;15(10):659-667.
doi pubmed - Cooke JP, Youker KA. Future Impact of mRNA Therapy on Cardiovascular Diseases. Methodist Debakey Cardiovasc J. 2022;18(5):64-73.
doi pubmed pmc - Stern JA, White SN, Lehmkuhl LB, Reina-Doreste Y, Ferguson JL, Nascone-Yoder NM, Meurs KM. A single codon insertion in PICALM is associated with development of familial subvalvular aortic stenosis in Newfoundland dogs. Hum Genet. 2014;133(9):1139-1148.
doi pubmed pmc - Drogemuller M, Jagannathan V, Dolf G, Butenhoff K, Kottmann-Berger S, Wess G, Leeb T. A single codon insertion in the PICALM gene is not associated with subvalvular aortic stenosis in Newfoundland dogs. Hum Genet. 2015;134(1):127-129.
doi pubmed - Abdallah H, Toomey K, O'Riordan AC, Davidson A, Marks LA. Familial occurrence of discrete subaortic membrane. Pediatr Cardiol. 1994;15(4):198-200.
doi pubmed - Petsas AA, Anastassiades LC, Constantinou EC, Antonopoulos AG. Familial discrete subaortic stenosis. Clin Cardiol. 1998;21(1):63-65.
doi pubmed pmc - Parker LE, Landstrom AP. Genetic etiology of left-sided obstructive heart lesions: a story in development. J Am Heart Assoc. 2021;10(2):e019006.
doi pubmed pmc - Piacentini G, Marino B, Digilio MC. Familial recurrence of discrete membranous subaortic stenosis. J Thorac Cardiovasc Surg. 2007;134(3):818-819.
doi pubmed - Fatimi SH, Ahmad U, Javed MA, Shamim S, Ahmad R. Familial membranous subaortic stenosis: review of familial inheritance patterns and a case report. J Thorac Cardiovasc Surg. 2006;132(6):1484-1486.
doi pubmed - Gale AW, Cartmill TB, Bernstein L. Familial subaortic membranous stenosis. Aust N Z J Med. 1974;4(6):576-581.
doi pubmed - Urbach J, Glaser J, Balkin J, Rosenmann D, Levy R, Marin G, Vidne B. Familial membranous subaortic stenosis. Cardiology. 1985;72(4):214-217.
doi pubmed - Helgadottir A, Thorleifsson G, Gretarsdottir S, Stefansson OA, Tragante V, Thorolfsdottir RB, Jonsdottir I, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun. 2018;9(1):987.
doi pubmed pmc - Ewart AK, Morris CA, Ensing GJ, Loker J, Moore C, Leppert M, Keating M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci U S A. 1993;90(8):3226-3230.
doi pubmed pmc - Olson TM, Michels VV, Lindor NM, Pastores GM, Weber JL, Schaid DJ, Driscoll DJ, et al. Autosomal dominant supravalvular aortic stenosis: localization to chromosome 7. Hum Mol Genet. 1993;2(7):869-873.
doi pubmed - Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet. 2000;1:461-484.
doi pubmed - Micale L, Turturo MG, Fusco C, Augello B, Jurado LA, Izzi C, Digilio MC, et al. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet. 2010;18(3):317-323.
doi pubmed pmc - Morris CA. Williams syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1249/.
- Zhou J, Wu Y, Xu X, Zhang Y, Zhang X, Chen H, Zhuang J, et al. Identification and characterization of novel elastin gene mutations in eleven families with supravalvular aortic stenosis. Front Genet. 2022;13:1059640.
doi pubmed pmc - Yasuhara J, Garg V. Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl Pediatr. 2021;10(9):2366-2386.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.