

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Case Report
Volume 13, Number 6, December 2022, pages 398-404

Overlapping Phenotype of Adult-Onset ALPK3-Cardiomyopathy in the Setting of Two Novel Variants
Olga S. Chumakovaa, b, e ![]() , Natalia V. Milovanovac
, Natalia V. Milovanovac ![]() , Igor O. Bychkovc
, Igor O. Bychkovc ![]() , Ekaterina Y. Zakharovac
, Ekaterina Y. Zakharovac ![]() , Elena A. Mershinad
, Elena A. Mershinad ![]() , Valentin E. Sinitsind
, Valentin E. Sinitsind ![]() , Dmitry A. Zateyshchikovb
, Dmitry A. Zateyshchikovb ![]()
aMoscow Healthcare Department, City Clinical Hospital 17, 119620 Moscow, Russia
bE.I. Chazov National Medical Research Center for Cardiology, 121552 Moscow, Russia
cResearch Centre for Medical Genetics, 115522 Moscow, Russia
dMedical Research and Educational Center, Lomonosov Moscow State University, 119991 Moscow, Russia
eCorresponding Author: Olga S. Chumakova, Moscow Healthcare Department, City Clinical Hospital 17, 119620 Moscow, Russia
Manuscript submitted November 8, 2022, accepted November 22, 2022, published online December 1, 2022
Short title: Adult Case of Biallelic ALPK3-Cardiomyopathy
doi: https://doi.org/10.14740/cr1449
| Abstract | ▴Top |
Inherited cardiomyopathies (CMPs) are fairly common causes of morbidity and mortality, particularly, in young individuals. In substantial number of cases, only morphological diagnostic criteria cannot distinguish one CMP from another because of incomplete penetrance, advanced stage of the disease, or overlapping phenotypes. Genetic testing has become a mandatory tool for definite diagnosis that is required for family screening, individual prognosis, and personalized treatment strategy in routine practice. In parallel, accumulation of genotype-phenotype correlations, especially for rare genes, promotes the deciphering of underling molecular mechanisms and the development of targeting treatment of CMPs. Here we present an adult-onset case comprised morphological features of several CMPs: asymmetric left ventricle (LV) hypertrophy, severe systolic dysfunction, LV hypertrabeculation and restrictive physiology. Using next-generation sequencing, two novel variants (NM_020778.5:c.1958C>G:p.Ser653* and c.3491G>A:p.Arg1164Gln) in alpha-protein kinase 3 (ALPK3) gene were identified and confirmed with Sanger sequencing. The trans-position (location on different alleles) of identified ALPK3 variants was established by plasmid cloning method. The ALPK3 gene, encoding nuclear alpha-protein kinase 3, has only recently been associated with CMPs and there are still few clinical data on ALPK3 variant carriers. To date, only five affected individuals with adult-onset CMPs in the setting of biallelic variants of ALPK3 gene have been reported.
Keywords: Hypertrophic cardiomyopathy; Dilated cardiomyopathy; Alpha kinase 3; ALPK3; Novel variant; Biallelic; Genetic testing
| Introduction | ▴Top |
Cardiomyopathies (CMPs) are a heterogeneous group of disorders characterized by structural and functional abnormalities of the myocardium that are not explained solely by abnormal loading conditions, such as hypertension or valve disease, or coronary artery disease [1]. CMPs are no longer considered to be rare diseases [2]. They are important causes of premature death from arrhythmia, progressive heart failure (HF) and stroke [3]. In classification of CMPs, based on imaging, there are four classical morphological subgroups: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic and restrictive cardiomyopathy [1]. The majority of CMPs has a genetic predisposition (inherited CMPs) and increasing progress in understanding the genetic basis of disease creates a new paradigm for targeting treatment of CMPs [4]. Through genotype-phenotype studies it has become evident that inherited CMPs have a complex genetic basis: pathogenic variants (mutations) in different genes can be the cause of one phenotype and, on the other hand, variants in one gene can lead to various cardiac phenotypes [5]. Thus, making a definite diagnosis based solely on clinical examination can be challenge.
HCM is the most common CMP affecting up to 1:200 of general population [6] and defined by thickening of left ventricle (LV) wall ≥ 15 mm in adult probands [7]. Classically, HCM is associated with small LV cavity, hyperdynamic contraction and LV outflow tract obstruction. HF in HCM is usually associated with preserved LV ejection fraction (EF), but about 6-8% HCM patients develop impaired systolic function with left ventricular ejection fraction (LVEF) less than 50%. This condition is often referred to as end-stage HCM, hypokinetic HCM, or burned-out cardiomyopathy that is phenotypically overlapping with DCM and associated with worse outcomes [8-10].
Although pathogenic variants in genes encoding sarcomeric proteins are the most frequent causes of HCM accounting for approximately 30-65% of probands, variants in several non-sarcomeric genes have been shown to be disease-causing [11]. One of such genes is alpha-protein kinase 3 (ALPK3), located on chromosome 15q25.2 and consists of 14 exons. First evidence in humans that mutations in ALPK3 cause CMPs was reported in 2016 in five children presenting with severe DCM or mixed DCM/HCM phenotype in utero, at birth or early childhood. All of them carried biallelic homozygous protein-truncated mutations in ALPK3 [12]. Similar phenotypes were reported later in other small predominantly pediatric case series of biallelic ALPK3 carriers [13-18]. The most patients revealed additional musculoskeletal abnormalities, facial dysmorphism and cleft palate. Notably, survived children demonstrated a transition from DCM to HCM with some extent of improvement of cardiac function and symptoms. This phenomenon has not been described before and appears to be unique to biallelic ALPK3-associated CMP. In 2021, Lopes et al received robust evidence that presence of protein-truncated variants in one allele (heterozygous variants) of ALPK3 also cause CMP with HCM phenotype. This autosomal dominant ALPK3-associated HCM is characterized by relatively late onset (56 ± 15.9 years) compared to sarcomere-positive patients, extensive myocardial fibrosis, and a progression of HF [19]. Heterozygous missense variants in ALPK3, both rare and common, have been also associated with DCM and later HCM [20-24], but pathogenicity for them is more challenging to ascertain and causal effect remains to be confirmed. There is still lack of data about the carriers of variants in ALPK3. To date, 27 patients carrying biallelic ALPK3 variants have been described and only five of them had an adult-onset presentation of CMP (Supplementary Material 1, www.cardiologyres.org). Here we present one more adult-onset case comprised morphological features of several CMPs in the setting of two novel variants (protein-truncated and missense) in ALPK3.
| Case Report | ▴Top |
Clinical presentation
Patient (Turkic origin male of 38 years old) visited our outpatient department with complains on breathiness during his daily life and chest pain not related to physical activities. At his 19 years a single syncope during sport competition occurred. He was examined (the documentation is not available), “congenital heart disease” was diagnosed, sport activities were restricted. He stayed asymptomatic without any medications until this year when the above symptoms firstly appeared. The patient denied smoking, alcohol abuse and drug use. His height was 172 cm, weight 80 kg, body mass index 27 kg/m2. There was no visible skin, musculoskeletal, neurological abnormalities. Blood pressure was 127/79 mm Hg, heart rate 72 beats per minute, respiratory rate 16 per minute.
Family history did not reveal cases of sudden cardiac death (SCD) among relatives. Both parents (mother of 59 years and father of 63 years) are alive and asymptomatic but have not been cardiological evaluated. The patient has three healthy children (1, 10 and 15 years) and two asymptomatic brothers of 35 and 36 years old. One more brother died at his 20 years in a car accident. The paternal grandmother died at her 50 years with the presence of ascites.
Diagnosis
Hematology blood test was normal. In biochemistry there were mild elevation of creatinine and urea levels, significant elevation of alanine aminotransferase level and twice elevation of creatine kinase (CK). The other parameters were within normal range. Vessel ultrasound did not reveal signs of peripheral atherosclerosis or pathology of deep veins. On his electrocardiogram (ECG) there were significant depolarization and repolarization abnormalities (Fig. 1).
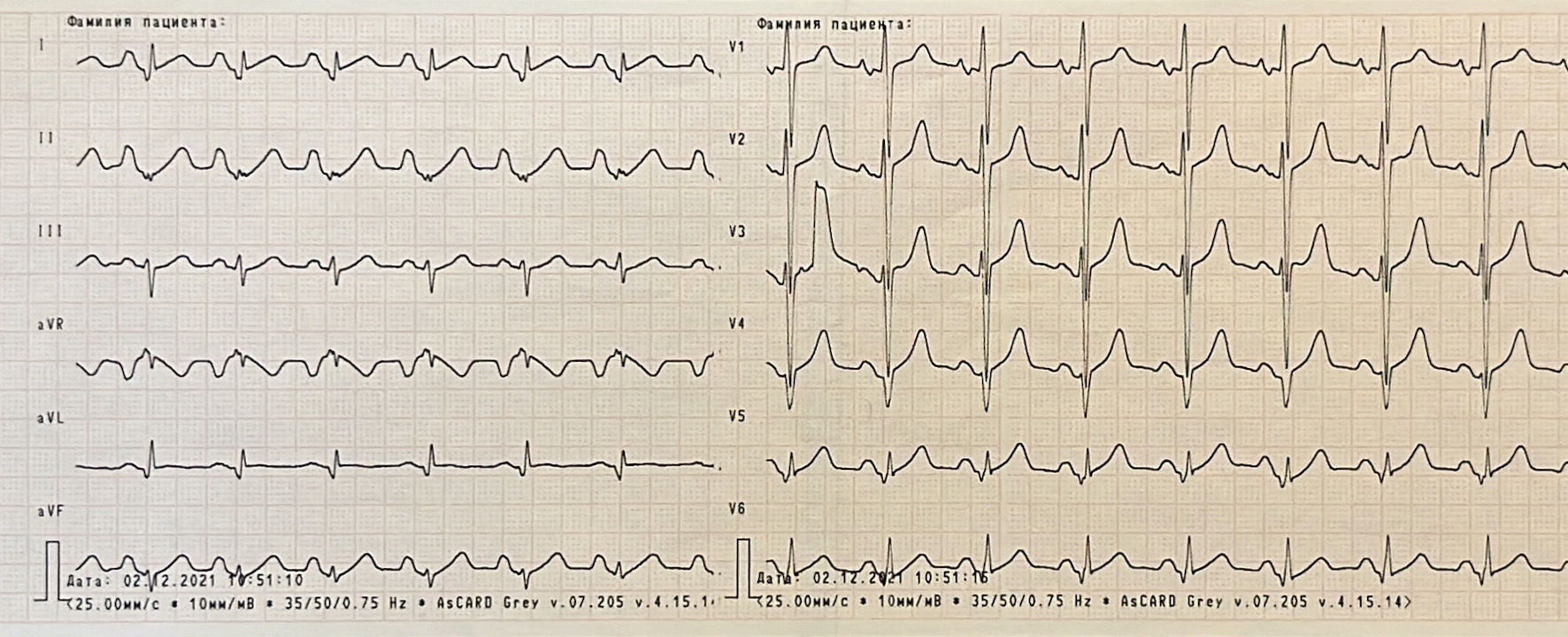 Click for large image | Figure 1. Electrocardiogram showing sinus rhythm, hypertrophy of both atriums and right ventricle, poor progression R in V4 - V6, pathological Q wave in lateral leads, ST segment elevation in I, V5 and V6, low QRS voltage in standard and V5 - V6 leads. |
Echocardiogram showed symmetric LV hypertrophy, severe impairment of LV systolic function (EF was 34%), restrictive type of LV diastolic function with pronounced left atrial enlargement and moderate pericardial effusion (Fig. 2a, b). Cardiac magnetic resonance (CMR) revealed LV hypertrophy with maximal LV wall thickness of 16 mm in basal and middle segments, excessive LV myocardial trabeculation, diffuse LV systolic dysfunction with EF of 30%, substantial subepicardial and transmural late gadolinium enhancement (LGE) in myocardial of both ventricles, left atriomegaly and moderate pericardial effusion. Pulmonary artery systolic pressure was 53 mm Hg. There was interatrial septal aneurysm (Fig. 2c, d).
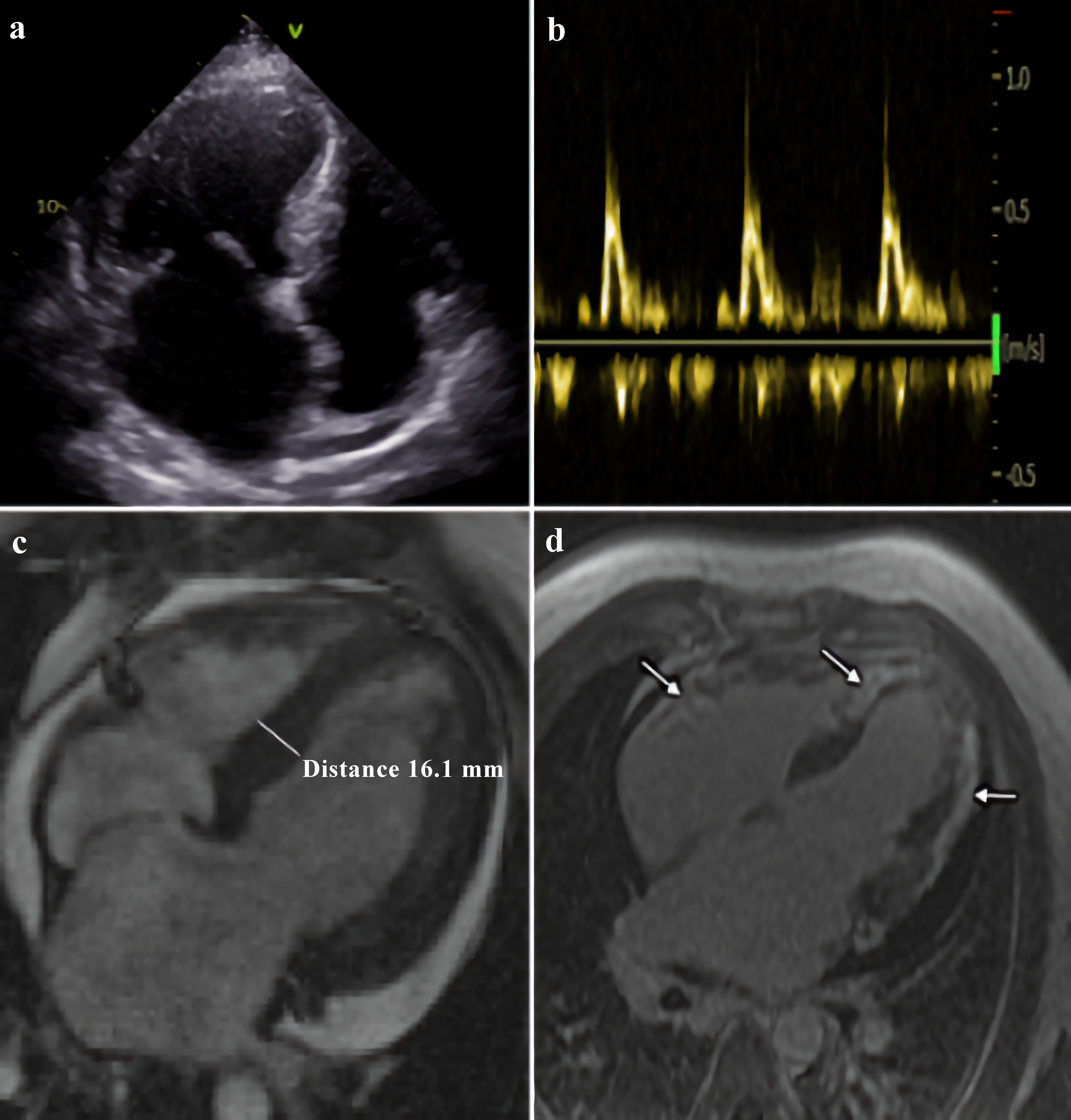 Click for large image | Figure 2. Morphological features of ALPK3-associated cardiomyopathy. Echocardiogram: apical four-chamber view (a), and transmitral flow, demonstrating restrictive type of LV diastolic function (b). Cardiac magnetic resonance: (c) LV hypertrophy in basal and middle segments, and (d) substantial late gadolinium enhancement in myocardial of both ventricles (showed by arrows). LV: left ventricle; ALPK3: alpha-protein kinase 3. |
Genetic testing using next-generation sequencing (NGS) method revealed two novel variants in ALPK3. One more variant in sarcomeric myosin-binding protein C (MYBPC3) gene was also found (Table 1). Sanger sequencing was performed to validate variants (Fig. 3).
 Click to view | Table 1. The Genetic Findings in This Case |
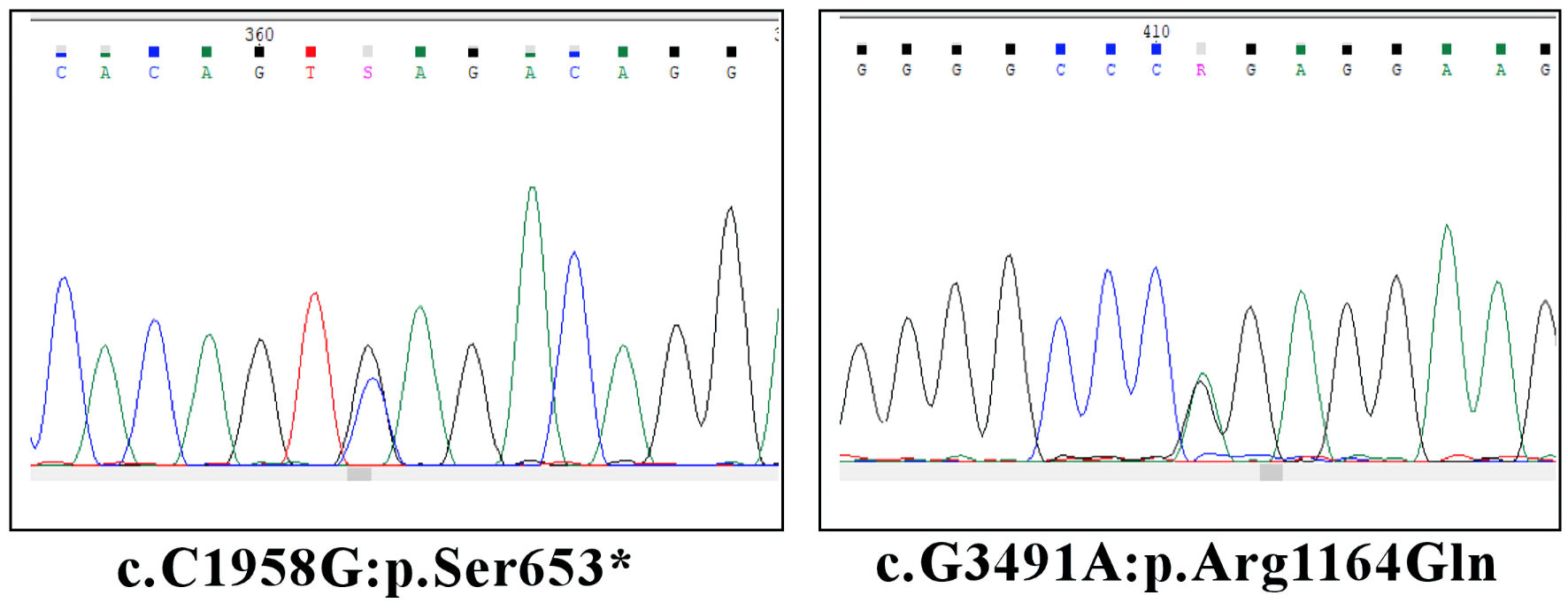 Click for large image | Figure 3. Validation of NGS ALPK3 variants by Sanger sequencing. ALPK3: alpha-protein kinase 3. |
Technical aspects of the genetic study
DNA was extracted from peripheral blood using the DNAPrep100 Kit (IsoGene, Moscow, Russia) according to the manufacturers’ recommendations and stored at -20 °C prior to analysis. Library preparation was performed using Parseq base module Prep&Seq™ U-target DNA kit according to manufactures’ recommendations. NGS was performed using the Ion S5 system (Thermo Fisher Scientific). A custom panel, compiled in Ion ParSeq Designer, covered transcribed sequences of 77 genes (exons and untranslated region (UTR) regions) was used (Supplementary Material 2, www.cardiologyres.org). Sequencing reads were aligned to the National Center for Biotechnology Information (NCBI) human reference genome (GRCh38/hg38). The obtained results were analyzed using Torrent Suite software. Integrative Genomics Viewer was used for performing visual analysis of the data and manual filtering of artifacts. Data analysis was performed with in-home NGSData [25]. The nomenclature of identified molecular variants were defined according to the Human Genome Variation Society guidelines (HGVS) [26]. For filtering of the reported variants according to their allele frequency the gnomAD database was used [27]. Human Gene Mutation Database (HGMD) [28] and National Center for Biotechnology Information [29] were used for searching previously described pathogenic variants. The set of bioinformatic tools (SIFT, SIFT4G, Polyphen2, MutationAssessor, FATHMM, PROVEAN, DEOGEN2, LRT, PrimateAI, MetaSVM, MetaLR, MutationTaster, SpliceAI, MMsplice, SpiP, Spidex) was used for predicting the pathogenicity of variants and the deleterious effects on protein. The recommendations of American College Medical Genetics (ACMG) were used for classification of novel variants. Sanger sequencing was performed using an ABI Prism 3500XL (Thermo Fisher Scientific, USA), following the manufacturer’s protocol.
Molecular cloning
To establish cis- or trans-position of identified ALPK3 variants, the fragment containing c.1958C>G (p.Ser653) and c.3491G>A (p.Arg1164Gln) variants was amplified from patient’s DNA by PCR with primers 5’-ATCAGAGCAGTTGTAGATGAGGTC-3’ and 5’-GAATCCCGAGGAGGGATATGCT-3’. The PCR product was ligated with pAL2-T vector (Evrogene, Russia) and transformed into competent XL10-Gold cells (Agilent, USA). Single colonies containing clones of a separate allele were collected, amplified by PCR and Sanger sequenced.
| Discussion | ▴Top |
Most of data regarding the clinical course of HCM were received from sarcomere-positive patients. There is extremely lack of data about non-sarcomere HCM patients, including ALPK3-causative ones. In our patient we found two novel variants in ALPK3.
It is known, that ALPK3 plays a role in heart development acting as a transcriptional regulator through the phosphorylation of cardiac transcription factors, such as HEY2, to promote cardiomyocyte differentiation and maturation [12, 30]. ALPK3 appears to be also necessary for normal intercalated disc formation and cardiomyofibril organization [12, 13]. ALPK3-mutant cardiomyocytes displayed significantly increased irregular calcium transients, indicative of abnormal calcium handling what has been observed in other human pluripotent stem cells (hPSC) models of HCM [13]. Recently it has been shown that in hPSC-derived cardiomyocytes modeling ALPK3 deficiency and cardiomyopathic ALPK3 mutations, sarcomeric organization is abnormal suggesting that ALPK3 has an integral role in maintaining sarcomere integrity and proteostasis in striated muscle [31].
In ALPK3-deficient mice, concentric cardiac hypertrophy of both ventricles was the predominant feature, but changes typically associated with DCM were also observed [32]. This is consistent with subsequent observations in humans, including our case.
Usually, HCM and DCM are described as opposite distinct entities, but in some cases, they may represent different stages of one pathological process. Our patient presented with overlapping HCM/DCM phenotype similar with most previous reported biallelic cases of ALPK3-related CMPs. Obviously, deficient of ALPK3 results in morphological features of both HCM and DCM in the same patients. Apart from pediatric cases, our patient demonstrated more benign course of the disease with onset in his middle age with mild symptoms. We presume that his first phenotype was HCM with further transformation to DCM. It is well known, that about 6-8% of HCM patients develop DCM phenotype (hypokinetic HCM) at the late stage of their disease. Among the factors responsible for such transformation, it has been reported clinical (early disease onset, greater wall thickness, family history of HCM [10], substantial LGE on CMR [33], atrial fibrillation (AF), cardiac surgery and device implantation [8]) and genetic (malignant mutations [34] or multiple pathogenic variants particularly in sarcomeric thin filament genes (TNNT2, TNNI3, TPM1, ACTC) [9]) factors. Based on our case together with the previous ALPK3 ones we can suggest one more risk factor for development of hypokinetic HCM - carrying variants in ALPK3.
First time biallelic ALPK3 variants in adults were described by Al Senaidi et al in 2019. There was a family with six affected members, two of them were asymptomatic adults with moderate isolated cardiac phenotype diagnosed at 38 and 28 years [16]. To our best knowledge, only 5 biallelic ALPK3 variants (together with two cases from Al Senaidi et al) in adult-onset cases have been described to date. There were both homozygous and compound heterozygous patients with truncating and missense variants (Supplementary Material 1, www.cardiologyres.org). Comparing to the pediatric cases, the phenotypic severity decreases with progression of age of onset. The clinical course of our patient is similar with five previously reported adult cases: asymptomatic or mild symptomatic middle-aged individuals with complex cardiac phenotype and without extracardiac manifestations. Looking at the families where the relatives-carriers of the same genotype demonstrate a wide range of ages of onset (from infancy to middle age), it is clear that additional genetic factors are involved in penetrance.
In our patient we identified novel protein-truncated pathogenic variant p.Ser653* in ALPK3 gene. The presence of this stop codon can lead to the formation of aberrant transcripts that can be degraded before translation by the cellular protein degradation machinery (generally the nonsense-mediated mRNA decay, or NMD, pathway) or be translated and generate truncated proteins. In the latter case, the protein could be degraded by lysosomal or ubiquitin-proteasome systems. This variant is located in exon 6. This is one of two biggest exons harboring the most variants previously associated with HCM. This variant is absent in gnomAD control populations. The causative role of the second novel missense variant p.Arg1164Gln in ALPK3 gene remains to be confirmed in the future modeling studies but the onset at early adult age and overt mixed phenotype testify in pathogenicity of this variant.
Our patient did not have pathogenic/likely pathogenic sarcomeric variants. Missense variant p.Glu223Lys in MYBPC3 is present in population databases (rs397516069, ExAC 0.1%) and has an allele count higher than expected for a pathogenic variant. It has been identified in one adult with HCM, however in that individual a pathogenic MYBPC3 variant on the other allele (in trans) was also identified. It is also well known that predominantly protein-truncated variants in MYBPC3 cause HCM. Despite MYBPC3 is one of two major HCM genes, this missense variant with borderline prevalence in general population could hardly play a key role in phenotype formation in our patient, but it may have a modifier role that is a challenge to determine for a while.
In cases where there are two variants in the same gene, determining the cis- or trans-position is crucial for further genetic counselling and assessment of risk disease transmission. In our case, the patient has transmitted one of two variants to all his children, which, even if genetic testing is not available for children, dictate careful monitoring of all of them.
In the present case the patient was mildly symptomatic but had severe morphological phenotype comprising extensive biventricular LGE and low LVEF that make his prognosis worse and dictate follow up him closely. According to American HCM SCD risk assessment model [35], our patient was judged to be at high risk of SCD and implantable cardioverter-defibrillator (ICD) implantation was recommended. However, the previous publications questioned the arrhythmic risk profile associated with ALPK3-CMP, particularly in autosomal dominant cases [19]. Thus, we should admit that there is a gap in our knowledge regarding risk of SCD in ALPK3-CMP patients and collecting data is mandatory for better decision-making. Hypokinetic restrictive phenotype with left atriomegaly makes our patient at a high risk of new onset AF and stroke. Thus, 24 - 48 h ECG Holter monitoring every 6 months was recommended as well. The protocol of clinical examination and genetic panel applied to exclude phenocopies of HCM in our patient.
Study limitations
We could not perform clinical and genetic examination of the parents of our patient and identify whether described variants are de novo or inherited, and in case of inheritance if heterozygous parents have clinical presentation of CMP. In light of the recently published data on the polygenic impact of many genetic variants into HCM, the role of other concomitant variants in the genes cannot be excluded and may need further elucidation. The rare deleterious missense variants were selected using prediction tools only, which carries a risk of false positives. Further studies on animal models and cardiomyocytes are needed to determine the pathogenicity of these variants.
Conclusions
We described one more case of ALPK3-associated CMP and identified two novel variants: heterozygous ALPK3 protein-truncated variant (p.Ser653*), which is associated with adult-onset HCM characterized by extensive fibrosis and hypokinetic restrictive physiology, and heterozygous missense variant (p.Arg1164Gln), which appears to contribute to the patient’s severe overlapping phenotype, creating compound heterozygosity. Further data collecting and modeling studies will allow developing a more personalized approach in this group of patients.
Learning points
Genetic testing offers opportunities for a better understanding and management of patients with CMPs and their families. Collecting and reporting clinical information about the patients harboring the disease-causing variants in novel and rare genes will expand our knowledge and promote the personalized treatment strategy in this population.
| Supplementary Material | ▴Top |
Suppl 1. Summary of patients with biallelic ALPK3-cardiomyopathy.
Suppl 2. The list of 77 genes in our custom panel associated with hypertrophic and restrictive cardiomyopathies, which was applied for genetic testing in presented case.
Acknowledgments
None to declare.
Financial Disclosure
The research was supported by the Russian Science Foundation (grant No. 20-15-00353).
Conflict of Interest
The authors declare that there is no conflict of interest.
Informed Consent
Patient consent for publication has been obtained.
Author Contributions
Conceptualization and study design: OSC (Olga S. Chumakova) and NVM (Natalia V. Milovanova); clinical investigation: OSC; genetic testing: NVM, IOB (Igor O. Bychkov) and EYZ (Ekaterina Y. Zakharova); cardiac imaging: OSC, EAM (Elena A. Mershina) and VES (Valentin E. Sinitsin); drafting of the manuscript: OSC, NVM and IOB; editing of the manuscript: EYZ and DAZ (Dmitry A. Zateyshchikov). Each author has reviewed the final version of the manuscript and approved it for publication.
Data Availability
The data that support the findings of this study are available within the manuscript.
Abbreviations
ACMG: American College Medical Genetics; AF: atrial fibrillation; ALPK3: alpha-protein kinase 3; CK: creatine kinase; CMP: cardiomyopathy; CMR: cardiac magnetic resonance; DCM: dilated cardiomyopathy; EF: ejection fraction; HGMD: Human Gene Mutation Database; HGVS: Human Genome Variation Society; HCM: hypertrophic cardiomyopathy; HF: heart failure; LGE: late gadolinium enhancement; LV: left ventricle; MYBPC3: myosin-binding protein C; NGS: next-generation sequencing; SCD: sudden cardiac death
| References | ▴Top |
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-276.
doi pubmed - McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol. 2021;18(1):22-36.
doi pubmed - Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L, Tendera M, et al. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J. 2018;39(20):1784-1793.
doi pubmed - Helms AS, Thompson AD, Day SM. Translation of New and Emerging Therapies for Genetic Cardiomyopathies. JACC Basic Transl Sci. 2022;7(1):70-83.
doi pubmed - Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 2019;140(7):595-610.
doi pubmed - Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254.
doi pubmed - Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-2779.
doi pubmed - Wasserstrum Y, Larranaga-Moreira JM, Martinez-Veira C, Itelman E, Lotan D, Sabbag A, Kuperstein R, et al. Hypokinetic hypertrophic cardiomyopathy: clinical phenotype, genetics, and prognosis. ESC Heart Fail. 2022;9(4):2301-2312.
doi pubmed - Marstrand P, Han L, Day SM, Olivotto I, Ashley EA, Michels M, Pereira AC, et al. Hypertrophic cardiomyopathy with left ventricular systolic dysfunction: insights from the SHaRe registry. Circulation. 2020;141(17):1371-1383.
doi pubmed - Biagini E, Coccolo F, Ferlito M, Perugini E, Rocchi G, Bacchi-Reggiani L, Lofiego C, et al. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol. 2005;46(8):1543-1550.
doi pubmed - Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. 2022;19(3):151-167.
doi pubmed - Almomani R, Verhagen JM, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, van der Zwaag PA, et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67(5):515-525.
doi pubmed - Phelan DG, Anderson DJ, Howden SE, Wong RC, Hickey PF, Pope K, Wilson GR, et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur Heart J. 2016;37(33):2586-2590.
doi pubmed - Caglayan AO, Sezer RG, Kaymakcalan H, Ulgen E, Yavuz T, Baranoski JF, Bozaykut A, et al. ALPK3 gene mutation in a patient with congenital cardiomyopathy and dysmorphic features. Cold Spring Harb Mol Case Stud. 2017;3(5):a001859.
doi pubmed - Jaouadi H, Kraoua L, Chaker L, Atkinson A, Delague V, Levy N, Benkhalifa R, et al. Novel ALPK3 mutation in a Tunisian patient with pediatric cardiomyopathy and facio-thoraco-skeletal features. J Hum Genet. 2018;63(10):1077-1082.
doi pubmed - Al Senaidi K, Joshi N, Al-Nabhani M, Al-Kasbi G, Al Farqani A, Al-Thihli K, Al-Maawali A. Phenotypic spectrum of ALPK3-related cardiomyopathy. Am J Med Genet A. 2019;179(7):1235-1240.
doi pubmed - Jorholt J, Formicheva Y, Vershinina T, Kiselev A, Muravyev A, Demchenko E, Fedotov P, et al. Two new cases of hypertrophic cardiomyopathy and skeletal muscle features associated with ALPK3 homozygous and compound heterozygous variants. Genes (Basel). 2020;11(10):1201.
doi pubmed - Herkert JC, Verhagen JMA, Yotti R, Haghighi A, Phelan DG, James PA, Brown NJ, et al. Expanding the clinical and genetic spectrum of ALPK3 variants: Phenotypes identified in pediatric cardiomyopathy patients and adults with heterozygous variants. Am Heart J. 2020;225:108-119.
doi pubmed - Lopes LR, Garcia-Hernandez S, Lorenzini M, Futema M, Chumakova O, Zateyshchikov D, Isidoro-Garcia M, et al. Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur Heart J. 2021;42(32):3063-3073.
doi pubmed - Aung N, Vargas JD, Yang C, Cabrera CP, Warren HR, Fung K, Tzanis E, et al. Genome-wide analysis of left ventricular image-derived phenotypes identifies fourteen loci associated with cardiac morphogenesis and heart failure development. Circulation. 2019;140(16):1318-1330.
doi pubmed - Pirruccello JP, Bick A, Wang M, Chaffin M, Friedman S, Yao J, Guo X, et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat Commun. 2020;11(1):2254.
doi pubmed - Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, Waring A, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53(2):135-142.
doi pubmed - Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, Kelu Bisabu K, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53(2):128-134.
doi pubmed - Dai J, Li K, Huang M, Sun Y, Liu H, Li Z, Chen P, et al. The involvement of ALPK3 in hypertrophic cardiomyopathy in East Asia. Front Med (Lausanne). 2022;9:915649.
doi pubmed - https://ngs-data.epigenetic.ru/main/.
- www.hgvs.org/mutnomen.
- http://gnomad.broadinstitute.org/.
- http://www.hgmd.cf.ac.uk.
- https://www.ncbi.nlm.nih.gov/.
- Hosoda T, Monzen K, Hiroi Y, Oka T, Takimoto E, Yazaki Y, Nagai R, et al. A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J Biol Chem. 2001;276(38):35978-35989.
doi pubmed - McNamara JW, Parker BL, Voges HK, Mehdiabadi NR, Bolk F, Chung JD, Charitakis N, et al. Alpha kinase 3 signaling at the M-band maintains sarcomere integrity and proteostasis in striated muscle. bioRxiv. 2022.
doi - Van Sligtenhorst I, Ding ZM, Shi ZZ, Read RW, Hansen G, Vogel P. Cardiomyopathy in alpha-kinase 3 (ALPK3)-deficient mice. Vet Pathol. 2012;49(1):131-141.
doi pubmed - Olivotto I, Maron BJ, Appelbaum E, Harrigan CJ, Salton C, Gibson CM, Udelson JE, et al. Spectrum and clinical significance of systolic function and myocardial fibrosis assessed by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. Am J Cardiol. 2010;106(2):261-267.
doi pubmed - Garcia-Giustiniani D, Arad M, Ortiz-Genga M, Barriales-Villa R, Fernandez X, Rodriguez-Garcia I, Mazzanti A, et al. Phenotype and prognostic correlations of the converter region mutations affecting the beta myosin heavy chain. Heart. 2015;101(13):1047-1053.
doi pubmed - Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020;76(25):e159-e240.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.