

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Review
Volume 13, Number 5, October 2022, pages 268-282

The Role of Fine Particles (PM 2.5) in the Genesis of Atherosclerosis and Myocardial Damage: Emphasis on Clinical and Epidemiological Data, and Pathophysiological Mechanisms
Aleksey Michailovich Chaulina, b, d ![]() , Artem Konstantinovich Sergeevc
, Artem Konstantinovich Sergeevc
aDepartment of Cardiology and Cardiovascular Surgery, Samara State Medical University, Samara 443099, Russia
bDepartment of Histology and Embryology, Samara State Medical University, Samara 443099, Russia
cDepartment of Hygiene, Samara State Medical University, Samara 443099, Russia
dCorresponding Author: Aleksey Michailovich Chaulin, Department of Cardiology and Cardiovascular Surgery, Samara State Medical University, Samara 443099, Russia
Manuscript submitted February 15, 2022, accepted August 5, 2022, published online October 25, 2022
Short title: PM 2.5 in Atherosclerosis and Myocardial Damage
doi: https://doi.org/10.14740/cr1366
- Abstract
- Introduction
- Description of PM 2.5
- Epidemiological and Clinical Research Data Providing Evidence of PM 2.5 Relationship With Atherosclerosis
- High Efficiency Particulate Air (HEPA) Filters and PM 2.5
- Pathogenetic Mechanisms Underlying the Proatherogenic Effect of PM 2.5
- Future Prospects and Research Directions
- Conclusion
- References
| Abstract | ▴Top |
Due to the fact that atherosclerotic cardiovascular diseases (CVDs) dominate in the structure of morbidity, disability and mortality of the population, the study of the risk factors for the development of atherosclerotic CVDs, as well as the study of the underlying pathogenetic mechanisms thereof, is the most important area of scientific research in modern medicine. Understanding these aspects will allow to improve the set of treatment and preventive measures and activities. One of the important risk factors for the development of atherosclerosis, which has been actively studied recently, is air pollution with fine particulate matter (PM 2.5). According to clinical and epidemiological data, the level of air pollution with PM 2.5 exceeds the normative indicators in most regions of the world and is associated with subclinical markers of atherosclerosis and mortality from atherosclerotic CVDs. The aim of this article is to systematize and discuss in detail the role of PM 2.5 in the development of atherosclerosis and myocardial damage.
Keywords: Cardiovascular diseases; Atherosclerosis; Particulate matter; PM 2.5; Pathogenesis; Endothelial dysfunction; Lipid metabolism; Oxidative stress; Inflammation; Autonomic nervous system
| Introduction | ▴Top |
Currently, a large number of risk factors and pathogenetic mechanisms underlying the development of cardiovascular pathologies are known [1-3]. However, all of them cannot fully explain all cases of cardiovascular diseases (CVDs) in humans. This indicates the presence of other unknown and potentially significant risk factors and mechanisms that need to be studied to optimize therapeutic and preventive measures. Recently, more and more attention has been paid to the study of associations of atmospheric air with CVDs [4-6].
The air that the population of the entire globe breathes is significantly polluted with numerous substances that can both increase the risk of development of various diseases in healthy individuals and lead to the aggravation of the existing pathologies [5-9]. Fine particulate matter (PM 2.5) constitutes one of the negative air pollutants. At the same time, it is especially alarming that only a small part of the population (about 18%) of our planet breathes the air in which the PM 2.5 content meets the normative indicators of the Guidelines on Air Quality (WHO, 2018) [9]. The recent “Global Burden of Diseases” report notes that exposure to the elevated PM 2.5 levels is the fifth leading risk factor for overall mortality in the population. PM 2.5 causes about 4.2 million deaths annually, the majority of which are the deaths from CVDs [10]. Atherosclerosis accounts for almost all known CVDs, and it is characterized by many risk factors for development and pathogenetic mechanisms, the study of which is being actively continued at the present time. Determining all risk factors and understanding the pathogenetic mechanisms of atherosclerosis is the essential step towards improvement of preventive and therapeutic measures and activities [11-14].
At present, many researchers regard PM 2.5 as the potential risk factor for the development of atherosclerosis and CVDs [14-17]. However, the potential pathogenetic mechanisms underlying the effect of PM 2.5 on the development and progression of atherosclerosis remain unclear.
Besides, several recent studies have found that the exposure to PM 2.5 leads to the damage of cardiomyocytes and the increase in the serum levels of cardiac biomarkers (high-sensitivity cardiac troponins, natriuretic peptides), inflammatory biomarkers (C-reactive protein, growth differentiation factor 15, fibrinogen, interleukin-6, interleukin-1 beta) and oxidative stress (8-OH-deoxyguanosine, catalase, malondialdehyde, nitrogen oxide, superoxide dismutase) [18-22]. Mechanisms of myocardial injury can be associated with both direct negative impact of PM 2.5 on myocardial cells and indirect impact (resulting from atherogenesis, which is accompanied by narrowing of coronary arteries, decrease in the delivery of oxygen and metabolic substrates to cardiomyocytes).
The aim of this article is to systematize and discuss in detail the role of PM 2.5 in the development of atherosclerosis and myocardial damage.
| Description of PM 2.5 | ▴Top |
Fine particulate matter constitutes the complex composition of solid particles and liquid droplets of microscopic size, which differ significantly in their make-up. Conventionally, we may distinguish two groups of sources of PM 2.5 formation: natural and human-driven (anthropogenic). The main natural sources of PM 2.5 air pollutants include volcanic eruptions, sandstorms, and plant sources. The sources of anthropogenic pollution are much more diverse: burning of fossil fuels, emissions from factories, smoke, road dust, vehicle exhaust gases, and a number of others. The specific make-up of PM 2.5 will differ in different regions of the world, since it depends on the sources of pollution and weather conditions, which may also significantly differ in different regions of the planet [23, 24]. This circumstance may also affect the physical and chemical properties of PM 2.5, the size of the particles, concentration and pathogenetic effect on the human body.
Depending on the aerodynamic diameter, they distinguish four types of PM: 1) large (PM 10) (diameter ≤ 10 µm), small (PM 2.5) (diameter ≤ 2.5 µm), PM 1.0 (diameter ≤ 1 µm) and ultrafine (diameter ≤ 0.1 µm). It is believed that the size of the particles that pollute the air determines the nature of their pathogenetic effect on the human body. Probably, the size of polluting particles is related to the ways and possibilities of penetration thereof into the certain organs and tissues of the human body. Thus, larger particles with the diameter in the range of 10 - 2.5 µm are mainly deposited in the nasal cavity, larynx, trachea, large and middle bronchi, which most often leads to the development of rhinitis, laryngitis, allergic diseases and bronchitis [25]. Smaller particles with the aerodynamic diameter in the range of 2.5 - 1.0 µm or less, can penetrate into smaller airways, such as small bronchi and alveoli, which can result in the development of inflammation directly in the lung tissue (pneumonia). The smallest (ultrafine) particles are so small that they can even pass through the alveolar-capillary barrier into the general blood circulation [26]. The concentration of PM 2.5 in the air determines the severity of the inflammatory response reaction and oxidative stress and positively correlates with the morbidity rate/incidence and mortality from CVDs across the population [27]. The characteristics and composition of PM 2.5 are also significantly influenced by the peculiarities of meteorological conditions, which can also be regarded as one of the factors determining the pathogenetic effect of PM 2.5. Thus, for example, organic components of PM 2.5 contribute to the development of atherosclerosis and CVDs, while inorganic components of PM 2.5 mainly affect the bronchopulmonary system [28]. Kim et al studied the relationship between the chemical components of PM 2.5 (sulfur, silicon, elemental carbon and organic carbon) and one of the main parameters of subclinical atherosclerosis - the carotid intima-media thickness (CIMT) in people living in six polluted urban areas of the United States for 2 years. The most significant increase in CIMT was specific to people who were exposed to organic carbon (0.026 mm (95% confidence interval (CI): 0.019 - 0.034) and a lower-value increase in CIMT was specific to people exposed to sulfur (0.022 mm (95% CI: 0.014 - 0.031)) and silicon (0.006 mm (95% CI: 0.000 - 0.012)) [29]. The results of another large study conducted by Sun et al confirm the connection of the chemical components of PM 2.5 with the development of subclinical atherosclerosis; however, the closest correlation with an increase in CIMT was specifically attributed to organic carbon [30]. Therefore, these studies are indicative of a different role of PM 2.5 chemical components in the pathogenesis of subclinical atherosclerosis. Moreover, the most significant role in the pathogenesis of atherosclerosis is played by organic chemical components of PM 2.5, in particular, organic carbon.
Despite the clear evidence of the relationship between the increased content of PM 2.5 in the air and the morbidity and mortality of the population from CVDs, the ways and mechanisms of PM 2.5 penetration into vascular cells and their direct toxic effects contributing to the development of atherosclerosis have been unknown for a long time. In light of this fact, several recent fundamental investigations have studied possible ways of PM 2.5 penetration into endothelial cells. Using human umbilical vein endothelial cells (HUVECs) as an object of research, the researchers demonstrated that the chemical components included in PM 2.5 can penetrate the cytoplasm of endothelial cells by four different mechanisms (Fig. 1) [31, 32]. The supposed mechanisms of entry are macropinocytosis, clathrin- and caveolin-mediated endocytosis [31]. At the same time, the accumulation of PM 2.5 components in umbilical vein endotheliocytes is not a harmless phenomenon, since it is accompanied by damage to mitochondria and lysosomes [32]. A number of other studies have also proved that the penetration of PM 2.5 into other cells (neural cells, alveolar macrophages) causes damage to mitochondria and oxidative stress in these cells [33, 34]. Thus, this mechanism is likely to be responsible for the development of endothelial dysfunction, that plays an important role in the initiation of atherogenesis.
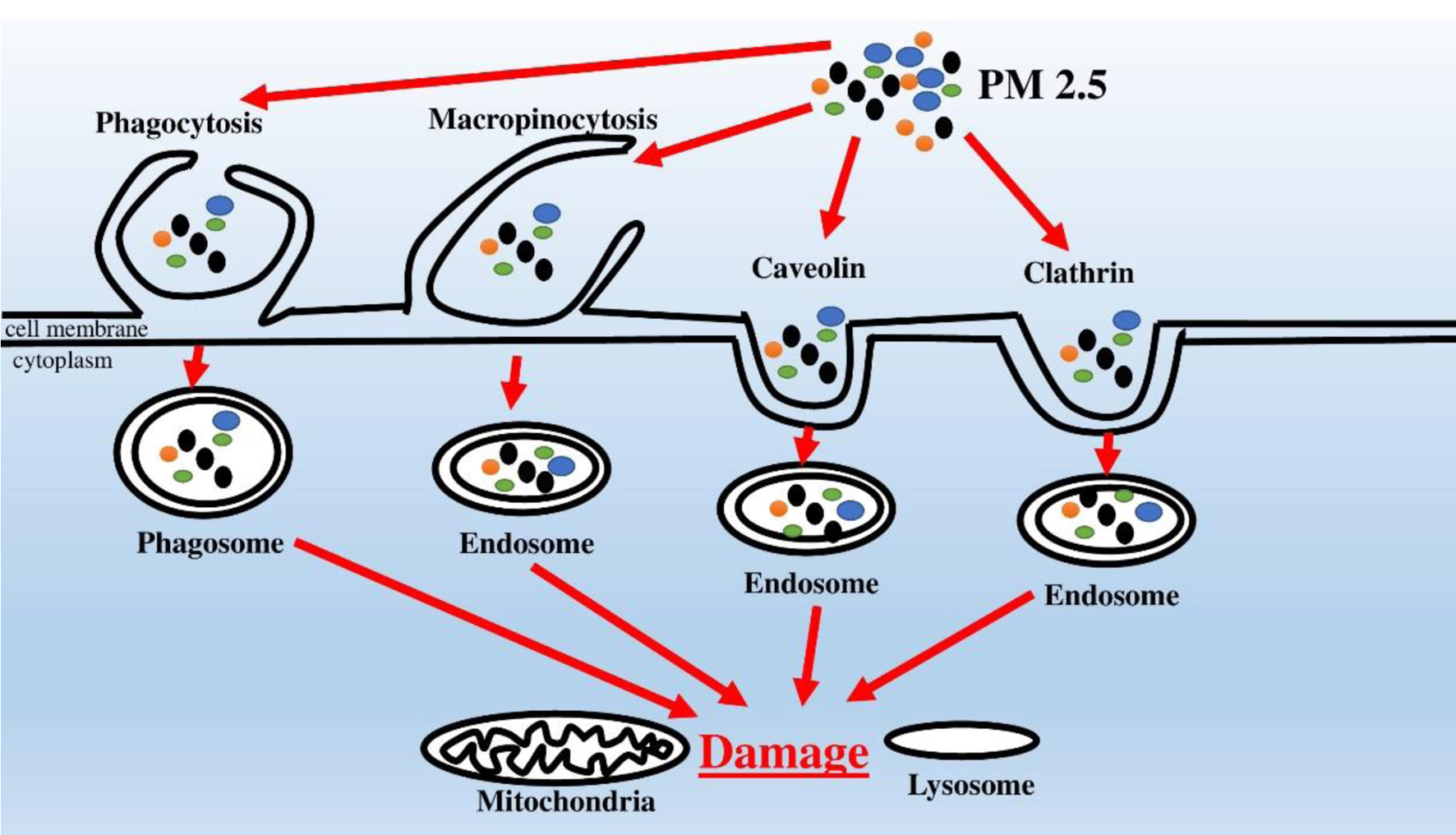 Click for large image | Figure 1. Mechanisms of PM 2.5 penetration into endotheliocytes and adverse intracellular effects. |
| Epidemiological and Clinical Research Data Providing Evidence of PM 2.5 Relationship With Atherosclerosis | ▴Top |
To date, the considerable amount of epidemiological and clinical research has been accumulated, confirming the adverse effects of PM 2.5 on human health, and in particular on the development and progression of atherosclerosis and CVDs. In many of these studies, subclinical parameters (markers) of atherosclerosis, such as CIMT, coronary artery calcification (CAC), thoracic aortic calcification (TAC) and ankle-brachialis index (ABI), have been assessed in healthy people living in disadvantaged regions. That said, almost all epidemiological researches carried out confirm the presence of a correlation between the exposure to PM 2.5 and these subclinical markers of atherosclerosis (Table 1) [35-44].
 Click to view | Table 1. Epidemiological Studies on the Relation of PM 2.5 With Atherosclerosis |
In one of the earliest epidemiological studies by Kunzli et al [35], researchers reported that a 10 µg/m3 increase in PM 2.5 was associated with the 5.9% increase in CIMT (95% CI: 1-11%). Notably, individuals of different gender and age were differently susceptible to the effects of PM 2.5. Thus, the highest degree of increase in CIMT (by 15.6%, 95% CI: 5.7-26.6%) was typical for women over 60 years old [35]. Subsequently, many studies have confirmed the existence of the relation between the exposure to PM 2.5 and the increase in the marker of subclinical atherosclerosis - CIMT [30, 36-39]. One of the significant sources of atmospheric air pollution (increased concentration of PM 2.5) is road transport, especially in conditions of heavy traffic. Hoffmann et al studied the relationship of prolonged stay of people in conditions of heavy traffic with one of the key markers of subclinical atherosclerosis (CAC), evaluated using electron beam tomography. According to the researchers, the most significant increase in CAC was typical for people living at a distance of less than 50 m from the road (1.63 (95% CI: 1.14 - 2.33)). As we moved away from the highway, the risk of subclinical atherosclerosis development is gradually decreased: 51 - 100 m - 1.34 (95% CI: 1.00 - 1.79), 101 - 200 m - 1 1.08 (95% CI: 0.85 - 1.39). According to the results of statistical analysis, when the distance between a residential building and a highway was reduced, there was the 7% increase in CAC (95% CI: 0.1 - 14.4) [40]. Another large prospective cohort study examined the relationship between prolonged exposure to polluted atmospheric air and the progression of coronary calcification and CIMT over 10 years of follow-up. Annually, the Cardiac Calcium Score (Agatston score) increased by 4.1 units (95% CI: 1.4 - 6.8) for each increase in the level of PM 2.5 by 5 µg/m3 in atmospheric air. However, in this study, exposure to PM 2.5 was not associated with progression of CIMT [41], which contrasts with the results of other academic specialists. The population-based cohort study conducted in the Ruhr region of Germany has shown that exposure to PM 2.5 is associated with the decrease in ABI, and it is in women that stronger associations were observed [42]. Kalsch et al investigated the relationship between PM 2.5 exposure and TAC, evaluated by electron beam tomography and considered a reliable marker of subclinical atherosclerosis. Prolonged exposure to fine particulate matter has been strongly associated with increased TAC [43]. Table 1 contains the main major clinical and epidemiological studies demonstrating a close relationship between elevated concentrations of PM 2.5 in ambient air (above the WHO recommended threshold of 10 µg/m3) and parameters of subclinical atherosclerosis (CIMT, CAC, ABI, and TAC).
A number of studies show the strong positive relationship between the elevated PM 2.5 levels and mortality from CVDs [44]. Crouse et al, in their largest cohort study involving 2.1 million non-immigrant Canadian adults, investigated the association between elevated PM 2.5 levels and cardiovascular mortality. Based on statistical analysis, the authors found that elevated concentrations of PM 2.5 (> 10 µg/m3) are associated with a higher risk of death from coronary heart disease (CHD) (1.31 (95% CI: 1.27 - 1.35) [45]. Along with that, the data obtained in the course of several studies indicate that even optimal PM 2.5 levels (< 10 µg/m3) in the ambient air may also increase the risk of adverse cardiovascular events. Thus, the studies conducted by Pinault et al [46] and Christidis et al [47] show the increased risk of mortality from CVDs at relatively low average PM 2.5 levels in the ambient air (at 6.3 and 5.9 µg/m3) [46, 47]. Huynh et al in their recent study found that low PM 2.5 concentrations (6.9 µg/m3) correlate with CAC grade in asymptomatic adult patients with low cardiovascular risk, regardless of other risk factors [48]. Thus, even an acceptable (by WHO standards) level of ambient air pollution is the risk factor for the development of atherosclerosis and the CHD. The results of these several studies indicate the need to adjust the safe values for PM 2.5 (< 10 µg/m3) recommended by the WHO Air Quality Guidelines. Further research is needed in this area to confirm the findings.
Lin et al found that long-term exposure to elevated concentrations of PM 2.5 is associated with key risk factors for the development of CVD (diabetes mellitus, hypertension, and obesity) in the Chinese population. Thus, each 10 µg/m3 increment of PM 2.5 was associated with increased prevalence of diabetes (odds ratio (OR): 1.118, 95% CI: 1.037 - 1.206), hypertension (OR: 1.101, 95% CI: 1.056 - 1.147), and overweight (OR: 1.071, 95% CI: 1.030 - 1.114) [49]. Ren and colleagues noted an association between short-term exposure to elevated PM 2.5 levels and the risk of hospitalization for cardiovascular and respiratory diseases [50]. A Chinese nationwide cohort study showed a close association between prolonged exposure to high atmospheric levels of PM 2.5 (46 ± 22 µg/m3) and the risk of CVD mortality. The main components of PM 2.5 (black carbon, organic matter, nitrate, ammonium, and sulfate) were formed as a result of burning fossil fuels [51].
Several studies have reported that targeted measures to prevent pollution and reduce PM 2.5 air concentration can effectively reduce the risk of atherosclerosis and CVDs in healthy population. Thus, the researchers found that alongside the decrease in the level of air pollution in Beijing, the serum concentrations of biomarkers of inflammation and oxidative stress in the surveyed population decreased [52, 53].
Thus, a number of clinical and epidemiological studies have clearly confirmed the existence of a close relationship between the concentration of PM 2.5 in the ambient air and subclinical markers of atherosclerosis, which allows PM 2.5 to be regarded as the significant and independent risk factor for the development and progression of atherosclerosis.
| High Efficiency Particulate Air (HEPA) Filters and PM 2.5 | ▴Top |
Taking into account the pathological role of polluted atmospheric air, reducing the concentration of PM 2.5 can be considered as one of the key therapeutic and preventive measures. Currently, HEPA filters are the most effective way to reduce the degree of atmospheric air pollution. HEPA filters are used in vacuum cleaners, air purification systems and ventilation and air conditioning systems [54]. According to a number of recent studies, the use of HEPA filters helps to significantly reduce PM 2.5 concentrations and their effect on the human body [55-57]. Thus, the use of a HEPA filter is an important method of combating PM 2.5, especially for people living in highly polluted regions of our planet.
| Pathogenetic Mechanisms Underlying the Proatherogenic Effect of PM 2.5 | ▴Top |
The main, but not the only mechanisms for the development and progression of atherosclerosis are oxidative stress, inflammation, endothelial dysfunction, lipid metabolism disorders, hemostasis disorders and impaired functioning of the autonomic nervous system (ANS). Since the formation of atherosclerotic plaques is an extremely complex and multistage process, involving the interaction of a number of cell populations (endothelial cells, monocytes, macrophages, smooth muscle cells, T-lymphocytes, etc.) and mechanisms [58-60], for a more holistic and accurate understanding of the role played by PM 2.5 in the development of atherosclerosis, one should consider separately the specific effect of PM 2.5 on each of the above-mentioned components and mechanisms. Therefore, below we will sequentially consider the effect of PM 2.5 on the above-mentioned mechanisms underlying the proatherogenic effect of PM 2.5.
PM 2.5 and endothelial dysfunction
Endothelial dysfunction is one of the generally recognized initiating mechanisms underlying the development and progression of atherosclerosis [58]. Under normal conditions, vascular endothelial cells produce a number of regulatory biologically active compounds that cause narrowing and dilation of blood vessels, depending on the existing needs of the human body. Various physicochemical factors, including PM 2.5, increased blood pressure (BP), reactive oxygen species (ROS)/reactive nitrogen species (RNS), low-density lipoproteins (LDLs), and oxidized LDLs (ox-LDLs), can cause damage to the endothelial cells or changes in the endothelial permeability [59]. In addition, many chemical components included in PM 2.5 can directly penetrate into endotheliocytes and cause damage to intracellular organelles (lysosomes, mitochondria), causing endothelial dysfunction [31, 32]. As a result of endotheliocyte damage, the barrier function of the endothelium weakens, which leads to increased transportation of many components of blood plasma, including LDLs through the endothelial barrier into the subendothelial space, where these atherogenic particles are accumulated/oxidized, subsequently initiating the formation of an arterial sclerotic disease [60-62]. LDL particles accumulated in the vascular walls undergo oxidation by RNS produced by endothelial cells and macrophages, which leads to the formation of ox-LDLs, the accumulation of which impairs the function of endothelial cells, completing the vicious pathogenetic circle [58, 62].
In order to assess the state of the vascular endothelium, the following main non-invasive methods are used: the test with reactive vascular hyperemia, measurements of the initial diameter of the arteries, and flow-mediated dilation (FMD) [63, 64]. The large study has shown that long-term exposure of a human body to PM 2.5 disrupts the function of endothelial cells by decreasing FMD and also causes vasoconstriction (reduction in the original diameter of the arteries) [65]. Academic specialists suggest that PM 2.5-induced dysfunction of endothelial cells is mainly mediated by the indirect cytotoxic effects caused by the inflammatory cytokines and oxidative stress [66]. Fine PM 2.5 particles induce the generation of ROS and RNS in the endothelial cells of the human lung vessels, which leads to the disruption of the endothelial barrier and to the secretion of a large amount of pro-inflammatory cytokines [67]. Several recent studies have shown that PM 2.5 can directly damage the endothelial cells of the human umbilical vein, which can be regarded as the trigger for the development of atherosclerosis [68, 69]. After damage to the endothelium, LDLs penetrate into the vascular wall through the damaged area of the endothelial cells and are oxidized by ROS to ox-LDLs. Besides, PM 2.5 causes overexpression of cell adhesion molecules such as intercellular adhesion molecules 1 (ICAM-1) and vascular adhesion molecules 1 (VCAM-1) on the surface of the endothelial cells. These adhesion molecules recruit leukocytes, such as T cells and monocytes, and promote their migration through the endothelial layer into the subendothelial space, where they multiply and differentiate into macrophages [70]. Besides, PM 2.5 enhances the secretion by the endothelial cells of monocytic chemotactic protein-1 (MCP-1), platelet-derived growth factor (PDGF) and macrophage colony stimulating factor (M-CSF), which enhance the migration of monocytes to the focus of atherosclerotic lesion and activate their differentiation into macrophages. At the same time, PDGF stimulates the migration and proliferation of smooth muscle cells that migrate from the middle layer of the vascular wall to the intima, where they multiply and capture atherogenic lipid particles. Both macrophages and smooth muscle cells take up atherogenic lipoproteins to form foam cells. A large number of foam cells are deposited in the subendothelial layer of the inner lining of the artery, forming yellow lipid stripes (spots), which, as a rule, do not rise, or slightly rise above the surface of the endothelium, which is the earliest sign of the development of atherosclerosis. Over time, the foam cells die, releasing their contents and thereby replenishing the contents of the necrotic nucleus of the atherosclerotic lesion focus, filled with lipids and cellular debris [58, 62].
Against the background of the accumulation of LDLs and ox-LDLs in the vascular wall conditioned by the endothelial dysfunction, on the surface of macrophages, the expression of scavenger receptors increases, which enhances the phagocytosis of LDLs and ox-LDLs, leading to the formation of foam cells [71]. Geng et al in their experimental study found that the effect of PM 2.5 on the bodies of experimental mice suffering from atherosclerosis stimulates the transformation of macrophages and smooth muscle cells into foam cells through the intracellular molecular pathway (TLR4/MyD88/NF-kB) (Fig. 2) [72]. This mechanism increases the risk of rupture of atherosclerotic plaques, which can contribute to its further growth inside the coronary artery lumen and increase the risk of myocardial infarction.
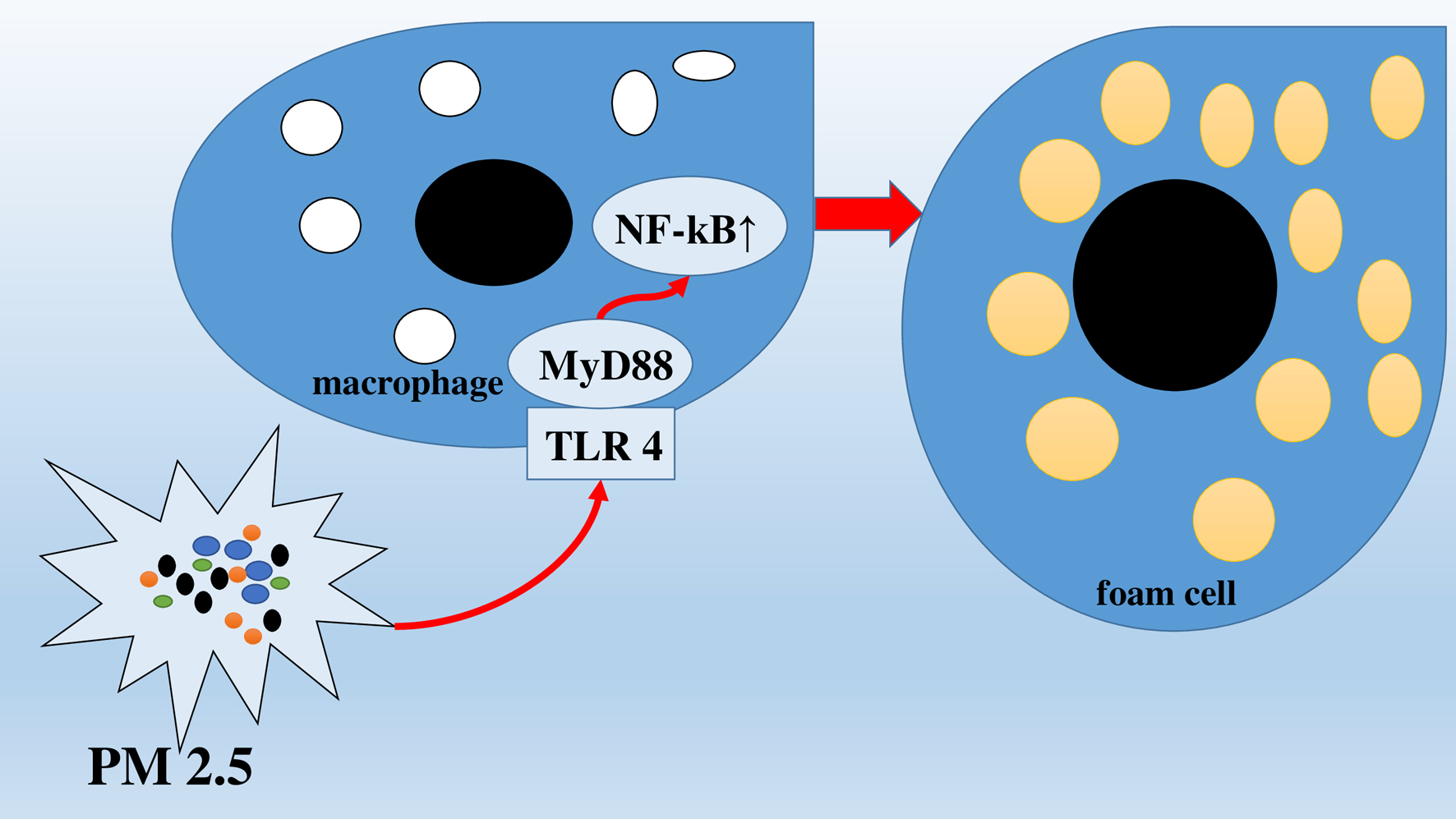 Click for large image | Figure 2. Transformation of macrophages into foam cells via intracellular molecular pathway (TLR4/MyD88/NF-kB). |
Besides, excess ox-LDLs content, conditioned by the effect of PM 2.5, stimulated the expression of the ox-LDL (LOX-1) receptor, enhancing the dysfunction of the endothelial cells and accelerating atherogenesis [73]. Thus, PM 2.5 can trigger and enhance atherogenesis due to the damage to and dysfunction of endothelial cells and participation in a number of further links in the pathogenesis of atherosclerosis (Fig. 3).
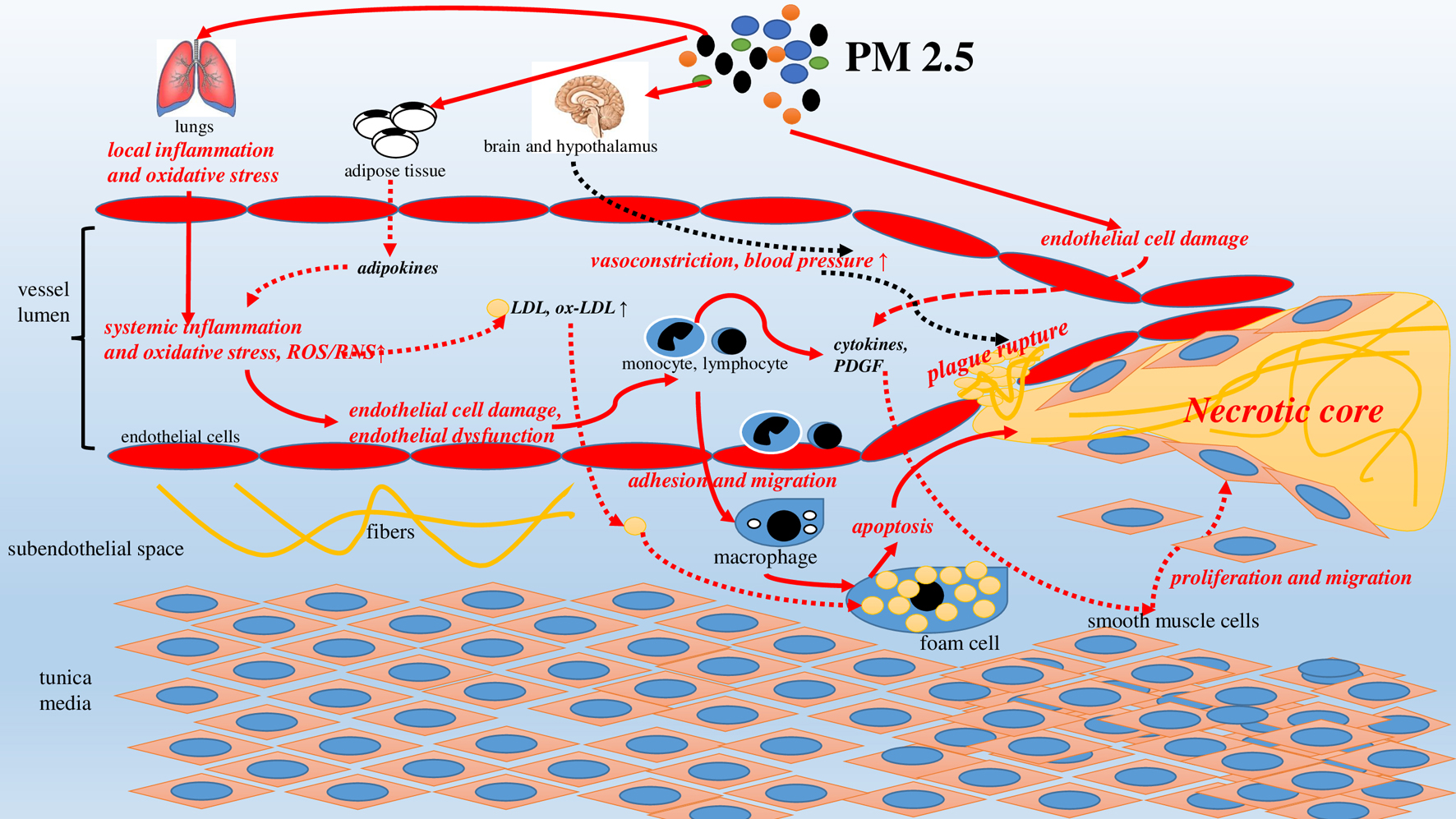 Click for large image | Figure 3. PM2.5-induced atherosclerosis: pathophysiological mechanisms. |
PM 2.5 and lipid metabolism disorders
More than 100 years ago, Russian academic specialists Anichkov and Ignatovskii were the first to show that lipid metabolism disorders are one of the key risk factors for the development of atherosclerosis and CVDs [74]. Although the dietary habits of people make the greatest contribution to the disturbance of lipid metabolism [74], some academic specialists have also traced the relationship between the effect of PM 2.5 fine particles and lipid metabolism disorders [75-77]. Thus, the randomized double-blind study has shown that exposure to PM 2.5 causes significant changes in the concentrations of a number of metabolic parameters of blood serum, in particular, the lipid spectrum, amino acids, glucose and several hormonal parameters [77]. The experimental study by Guan and colleagues has shown that the exposure of adult rats to PM 2.5 for 12 weeks leads to apparent changes in lipid profile (increase in the level of triglycerides, LDLs, and cholesterol). Adverse changes in lipid profile have been accompanied by atherosclerotic CVD [78].
A number of other studies have also clearly shown that exposure to PM 2.5 on the human body can cause dyslipidemia due to the increase in the concentration of total cholesterol, LDLs, triglycerides, and also lowering of antiatherogenic lipid (high-density lipoproteins, or HDLs) levels, which, in its turn, will increase the risk of formation of atherosclerotic plaques [79, 80]. The Multi-Ethnic Study of Atherosclerosis Air Pollution (MESA) has shown that HDL concentration in the blood serum decreases by 0.64 µmol/L (95% CI: -0.82 to 0.71 µmol/L) for each increase in the concentration air pollutants PM 2.5 of 5 µg/m3 [81]. Thus, the adverse effect of PM 2.5 negatively correlates with the level of antiatherogenic (protective) HDLs in the human blood serum.
Several recent studies have reported that functional properties of HDLs are no less important than their serum levels. Chemical substances contained in PM 2.5 enter into oxidation-reduction reactions with protein and lipid components of HDLs causing the dysfunction of the latter [82]. Another study has also found that short-term exposure to high PM 2.5 levels reduced the antioxidant and anti-inflammatory properties of HDLs [83]. Specific antiatherogenic effects of HDLs that are impaired by the effect of PM 2.5 include restricting the transport of excess cholesterol to the subendothelial space and macrophages, increasing the breakdown of oxidized phospholipids and ox-LDLs, and inhibiting LDLs oxidation [84, 85]. In the pilot study, long-term exposure to elevated PM 2.5 levels was associated with increased cholesterol and macrophage levels in the atherosclerotic plaques in case of laboratory mice [86]. Compared to the mice exposed to the air cleaned of PM 2.5, the mice exposed to PM 2.5 had decreased HDLs levels and increased levels of LDLs and ox-LDLs [87]. It has also been shown that PM 2.5 can enhance the development of atherosclerosis by increasing the expression of scavenger receptors and the accumulation of the oxidatively modified form of cholesterol (7-ketocholesterol) in the vascular wall [88]. Besides, PM 2.5 can also lead to the disorder of lipid metabolism due to the changes in the composition of the intestinal microflora, which is associated with the progression of atherosclerosis [89]. Thus, these data provide compelling evidence that PM 2.5 can accelerate the accumulation of lipids in the vascular wall, the formation of atherosclerotic plaque and its growth by altering the lipid metabolism: increasing the level of atherogenic and decreasing the level of anti-atherogenic particles, stimulating the oxidation of LDLs and phospholipids, changing the expression and functioning of scavenger receptors, LDLs receptors and ox-LDLs receptors.
PM 2.5, oxidative stress and inflammation
PM 2.5 contained in the air, during inhalation, enter the bronchial tree and alveoli and are deposited on their walls. These foreign particles trigger first local and then systemic immune-inflammatory reactions, which is accompanied by cell damage, generation of ROS and increased oxidative stress. Oxidative stress and immune-inflammatory reactions are one of the key links in the pathophysiology of atherosclerosis both at the initial stage of its development and at subsequent stages of the pathogenesis of atherosclerosis [58-62].
Chemical constituents of PM 2.5 depend on many factors and is very diverse. The most frequent components of PM 2.5 are organic substances (aldehydes, polycyclic aromatic hydrocarbons, organic carbon, etc.) and inorganic compounds (transition and heavy metal ions, silicon, sulfur, elemental carbon, etc.), forming a complex mixture. When these substances make it into the body, they can alter redox homeostasis and redox processes due to the intensification of the ROS/RNS formation [90]. The accumulation of the latter leads to oxidative stress and damage to the structural components of cells, which, in its turn, induces the inflammatory response reaction. Oxidative stress and inflammatory processes are closely interrelated and potentiate each other’s action, leading to the damage of the structural components of cells. Thus, oxidative stress is accompanied by increased formation of ROS/RNS, which cause direct damage to the structure of DNA, lipids and protein molecules that make up intracellular organelles and cell membranes. Damage to organelles and cell membranes can lead to impaired cell function, cell death and the release of cytoplasmic contents into the extracellular space, which can enhance local immune-inflammatory responses [58]. In addition, ROS and RNS stimulate the secretion of various inflammatory cytokines and mediators by immune and non-immune cells and promote the oxidation of LDLs in the blood to ox-LDLs, as well as stimulate the secretion of chemokines and pro-inflammatory factors by endothelial cells, which will further worsen the redox homeostasis in the body and form the vicious pathogenetic circle [58, 62, 91]. Heavy metal ions loaded on the surface of PM 2.5 can catalyze the Fenton reaction to produce ROS. Elevated levels of ROS enhance the activity of the nicotinamide adenine dinucleotide phosphate oxidase enzyme (NADPH oxidase), which causes mitochondrial damage and cell dysfunction [92]. A recent study has shown that PM 2.5 can cause mitochondrial damage in macrophages, activate the mitochondrial apoptosis pathway, increase lipid accumulation therein, and induce apoptosis of foam cells and macrophages, which further aggravates the pathogenesis of atherosclerosis [93].
The bronchopulmonary system is directly exposed to various air pollutants, including PM 2.5. PM 2.5 adsorbed on the surface of the airways can damage the epithelial cells of the airways and initiate local inflammation of the bronchi and alveoli (bronchitis, alveolitis), which leads to the production of a large amount of inflammatory mediators and ROS (Fig. 3). Given the high degree of blood supply to the bronchopulmonary system, inflammatory mediators and ROS in large quantities will penetrate into the systemic inflammatory blood flow, contributing to the development of the systemic inflammatory response [94]. In addition, a recent study has shown that in addition to mechanical damage to cells, PM 2.5 can directly activate the MyD88/NF-kB signaling pathway through Toll-like receptors (TLRs) on the surface of alveolar macrophages in the lungs. Effects of PM 2.5 on several types of TLRs, in particular TLR2 and/or TLR4, enhance the release of inflammatory cytokines by macrophages [95]. The systemic inflammatory response results in the significant increase in the concentration of pro-inflammatory factors, leading to the increased migration and proliferation of monocytes, migration of smooth muscle cells, and transformation of monocytes, macrophages and smooth muscle cells into foam cells, which promotes the formation and growth of the atherosclerotic plaque.
Several studies indicate the possible role of PM 2.5 components in inflammatory processes taking place in certain organs and tissues, among which the adipose tissue is of great importance [96-99]. Thus, in case of prolonged exposure, the toxic components of PM 2.5 by means of the bloodstream penetrate the adipose tissue, where they accumulate in large quantities and cause chronic inflammation and the release of adipokines into the bloodstream. Many of these adipokines, including visfatin, resistin and adiponectin, are directly involved in the pathogenesis of atherosclerosis by way of regulating the functioning of endothelial, smooth muscle cells and macrophages [100-103]. In general, the inflammatory response and oxidative stress are mutually supportive and interacting to induce a number of atherogenesis processes from formation to rupture of atherosclerotic plaques [57, 62]. Thus, PM 2.5 contributes to the onset and development of atherosclerosis by inducing the oxidative stress and the inflammatory response reaction.
PM 2.5 and the ANS
Although the ANS plays a vital role in the regulation of the cardiovascular system (CVS), its excessive activation under certain physiological and pathological conditions adversely affects the functioning of the CVS and can contribute to the development and progression of many CVDs [104, 105]. The main markers for assessing the state of the ANS are heart rate (HR), heart rate variability (HRV), and BP [106, 107]. Interestingly, the impact of PM 2.5 can lead to dysfunction of the ANS and can change the above-mentioned parameters, which is fraught with the formation of CVDs. Thus, several epidemiological studies have shown that exposure to PM 2.5 can disrupt the functioning of the ANS, in particular, lead to the changes in the HR and HRV [106, 107]. Several clinical and experimental studies have shown that excessive oxidative stress and systemic inflammation induced by PM 2.5 are accompanied by changes in the functioning of the ANS, which is expressed by the increase in BP and HR, and by the decrease in HRV [108-110]. Exposure to PM 2.5 can also result in dysfunction of the sympathetic nervous system, causing rapid changes in BP regulation. Thus, Fuks et al in their population-based cohort study (n = 4,539) showed that prolonged exposure to elevated concentrations of PM 2.5 in residential premises is associated with increased BP and a higher risk of arterial hypertension (AH) over the next 5 years of follow-up [111]. According to the Multi-Ethnic Study of Atherosclerosis (MESA), BP was positively associated with PM 2.5. Thus, an increase in the 30-day average concentration of PM 2.5 by 10 µg/m3 was associated with an increase in BP by 1.12 mm Hg (95% CI: 0.28 - 1.97). In addition, the association between PM 2.5 and BP was stronger in the presence of higher traffic exposure [112].
Animal experiments have shown that exposure to PM 2.5 can lead to direct damage and inflammation of the hypothalamus, which, in its turn, may be associated with over-activation of the sympathetic nervous system [113]. Another study reports that PM 2.5 impairs the functioning of the parasympathetic nervous system by way of increasing the methylation of the genes encoding the production of the pro-inflammatory cytokines [114]. Thus, PM 2.5 can directly disrupt the functioning of the ANS and increase the risk of AH. AH, in its turn, is the independent risk factor for the formation and aggravation of atherosclerosis and CVDs by way of a number of known mechanisms [115-120]. The increased level of BP in itself can damage the endothelium as a result of the hemodynamic shock and activation of the oxidative stress, which leads to the increase in the synthesis of collagen and fibronectin by the endothelial cells. AH can also result in the activation of the enzymes that causes oxidation of LDLs and the increase in the formation of ox-LDLs [119]. In case of AH, there is a higher expression of PDGF, which additionally induces the migration and proliferation of smooth muscle cells in the intima of the vessels, making atherosclerosis worse (Fig. 3) [111]. Besides, high BP is the risk factor for the rupture of atherosclerotic plaques and further adverse consequences in the form of acute cardiovascular accidents (acute myocardial infarction, sudden cardiac death, stroke, etc.) [121-123]. This is due to the fact that after the rupture of the atherosclerotic plaque, its contents are released into the bloodstream, in particular inflammatory and prothrombotic factors (collagen, tissue factor, thromboxane, etc.) that trigger the process of thrombus formation, as well as the fragments of the atherosclerotic plaque that cause rapid mechanical obstruction of the blood vessels (coronary or intracerebral), into which they get.
Impact of PM 2.5 on the hemostatic system
The exposure of humans and experimental animals to PM 2.5 increases thrombogenicity and thrombosis, which is indicative of quite a significant impact of PM 2.5 on the hemostatic system [124-128]. Thus, the experimental study by Nemmar et al has found that the exposure to exhaust particles increases platelet activation and causes peripheral thrombosis [124]. Some inflammatory mediators released into the blood stream on exposure to pollutants can accelerate processes of blood coagulation. Mutlu and colleagues reported that the exposure of laboratory mice to PM 2.5 enhances the production of interleukin-6 by alveolar macrophages. This inflammatory mediator, in its turn, increases the level of fibrinogen, the activity of coagulation factors II, VIII, and X of the hemostatic system, which leads to reduction of coagulation time and accelerated formation of arterial thrombus [125]. Experimental studies are consistent with the results of clinical studies. Under the exposure of a healthy human body to PM 2.5, there can be observed a reduction of prothrombin time, activated partial thromboplastin time and an increase in plasma levels of fibrinogen and D-dimers, which indicates a hypercoagulable state [126-128].
PM 2.5 and subclinical myocardial injury
A number of recent studies have shown evidence that PM 2.5 causes subclinical damage of cardiac muscle tissue [18-22]. Thus, even short-term exposure to environmental pollutants, including PM 2.5, leads to a significant increase in serum levels of high-sensitivity cardiac troponin I (on average by 22.9-154.7% compared to initial concentrations) in people living in contaminated areas [18]. In addition to changes in the concentration of troponins, the examined people showed increased levels of growth differentiation factor 15 and 8-OH-deoxyguanosine, which indicated the enhancement of inflammatory processes and oxidative stress [18]. The results of this clinical study are consistent with the results of the experimental study. Laboratory animals exposed to atmospheric air pollutants had significantly higher serum concentrations of the cardiac marker (cardiac troponin I), the inflammatory marker (C-reactive protein) and oxidative stress markers (malondialdehyde, catalase, nitrogen oxide, and superoxide dismutase), compared with the control group (P < 0.05) [22].
In general, high-sensitivity cardiac troponins are not only valuable biomarkers of the acute myocardial infarction, they also allow detecting myocardial injury in other pathological and physiological conditions that have a negative effect on cardiomyocytes [104, 118, 129-134]. The mechanisms of the increase of cardiac troponins in blood serum under the exposure of a human body to PM 2.5 are not definitely known. Basing on the analysis of the results of a number of clinical and experimental studies [18-22], there can be distinguished several mechanisms underlying the PM 2.5-induced myocardial injury and the increase of cardiac troponins in blood serum: 1) Initiation of cardiomyocyte apoptosis; 2) Enhancement of oxidative stress accompanied by increased production of ROS that damage cell membranes of cells, including cardiomyocytes. Damage of cell membrane of myocardial cells may be accompanied by an increase in the release of cardiac troponin molecules localized in the cytoplasm (cytosolic fraction of cardiac troponin). This fraction of troponins contains relatively few (approximately 5%) molecules of their total amount in the cardiomyocyte, hence, the degree of increase in serum levels of cardiac troponins under the exposure to PM 2.5 is also relatively small; 3) Subclinical ischemic injury caused by narrowing of the lumen of coronary vessels due to the development and progression of atherosclerosis. Thus, high-sensitivity cardiac troponins can be considered as laboratory biomarkers for assessing myocardial injury and the state of the cardiovascular system in people living in highly contaminated regions, which needs further research and clarification.
| Future Prospects and Research Directions | ▴Top |
Despite the abundance of established associations of PM 2.5 with subclinical markers of atherosclerosis [30, 35-43], CVD risk factors [49], the risk of hospitalization for CVDs [50] and an unfavorable prognosis [51], the specific pathogenetic mechanisms underlying the adverse effects of PM 2.5 on the CVS remain poorly understood. The chemical composition of PM 2.5 is an important factor that can significantly affect the pathogenesis of atherosclerosis and CVDs. This is due to the fact that different components of PM 2.5 have different effects on the inflammatory process, oxidative stress, lipid metabolism disorders and the hemostasis system. Thus, according to Huang et al among the numerous components of PM 2.5, only sulfates, iron and selenium were associated with the degree of inflammatory response (increased percentage of neutrophils in bronchoalveolar lavage) [135]. Transition metal ions (iron, copper, manganese and vanadium) can catalyze the Fenton reaction and similar reactions, which leads to a significant increase in the formation of ROS (superoxide radical, hydrogen peroxide and hydroxyl radical) and a subsequent increase in oxidative stress [136-138]. Accumulation of the above-described PM 2.5 components in vascular cells (endothelial cells, smooth muscle cells and macrophages) will cause oxidative stress, which will further lead to disruption of the functioning of these cells, in particular endothelial dysfunction, inflammatory reaction, transformation of macrophages and smooth muscle cells into foam cells, followed by apoptosis of these cells, which will enhance the growth of necrotic nuclei of atherosclerotic plaque. In the future, this may be associated with the risk of adverse cardiovascular events, and in particular, myocardial infarction [139].
The influence of many individual components on the development of atherosclerosis and CVDs is contradictory according to various studies and the specific mechanisms are not definitively known [135-138]. This is due to the multicomponent composition of PM 2.5 in the real air environment and the presence of other factors in the population (lifestyle, time of exposure to PM 2.5, the presence of chronic subclinical diseases, etc.), which can also affect the pathogenesis of atherosclerosis and CVDs, so it is difficult to understand what effect is characteristic of each specific component. To uncover specific mechanisms, strictly controlled experimental and clinical studies are necessary with the exclusion of all potentially influencing factors. This will help to establish the specific impact of PM 2.5 for each unfavorable region and develop specific preventive and curative measures to reduce the negative impact of PM 2.5. For example, in those regions where PM 2.5 components have pro-inflammatory and pro-oxidant activity, the main recommendation for individuals (in addition to using HEPA filters [55-57]) will be taking anti-inflammatory and antioxidant drugs. Thus, the clarification of pathophysiological mechanisms is important for the reasoned appointment of therapeutic and preventive measures.
| Conclusion | ▴Top |
According to the conducted narrative review, PM 2.5 should be regarded as the significant risk factor for the development of atherosclerosis and CVDs. This is confirmed by the number of clinical and epidemiological studies that have revealed the existence of close relationship between the increase in the concentration of PM 2.5 in the ambient air and markers of subclinical atherosclerosis (CIMT, CAC, TAC, and ABI). The pro-atherogenic effect of fine particulate matter is based on several basic and closely interrelated pathophysiological mechanisms: endothelial dysfunction, impaired lipid metabolism, increased oxidative stress and inflammatory reactions, impaired functioning of the ANS and increased activity of the hemostatic system. In addition, PM 2.5 causes subclinical injury of cardiac muscle cells by several mechanisms: apoptosis, oxidative stress, decreased oxygen delivery due to coronary atherosclerosis and ischemic damage of cardiomyocytes. Highly sensitive cardiac troponins are promising markers for detecting subclinical myocardial damage in people living in polluted regions. Further studies are needed to clarify the specific pathogenetic mechanisms, which in the future will help to reasonably prescribe optimal preventive and therapeutic measures.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have a financial relationship with any commercial entity that has an interest in the subject of this manuscript.
Conflict of Interest
The authors declare that they do not have a conflict of interest.
Author Contributions
Both authors participated in review. They were involved in writing and revising the article prior to submission.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Rabe KF, Hurst JR, Suissa S. Cardiovascular disease and COPD: dangerous liaisons? Eur Respir Rev. 2018;27(149):180057.
doi pubmed - Chaulin AM, Karslyan LS, Bazyuk EV, Nurbaltaeva DA, Duplyakov DV. [Clinical and diagnostic value of cardiac markers in human biological fluids]. Kardiologiia. 2019;59(11):66-75.
doi pubmed - Chaulin A, Duplyakov D. Analytical review of modern information on the physiological and pathochemical mechanisms of the release of cardiospecific proteins from muscle tissue, methodology and technologies of their research, interpretation of the results. Laboratory Diagnostics. Eastern Europe. 2022;11(1):78-97. (in Russian).
doi - Zhao M, Hoek G, Strak M, Grobbee DE, Graham I, Klipstein-Grobusch K, Vaartjes I. A global analysis of associations between fine particle air pollution and cardiovascular risk factors: feasibility study on data linkage. Glob Heart. 2020;15(1):53.
doi pubmed - Chaulin AM, Duplyakov DV. The role of environmental factors in the pathogenesis of cardiovascular diseases Part 1. Air Pollution. Arhiv Euromedica. 2021;11(1):30-35.
doi - Bevan GH, Al-Kindi SG, Brook RD, Munzel T, Rajagopalan S. Ambient Air Pollution and Atherosclerosis: Insights Into Dose, Time, and Mechanisms. Arterioscler Thromb Vasc Biol. 2021;41(2):628-637.
doi pubmed - Chaulin AM, Duplyakov DV. Cardiovascular diseases and chronic obstructive pulmonary disease: etiopathogenetic relationship and clinical significance (literature review). The Siberian Journal of Clinical and Experimental Medicine. 2020;35(2):26-34. (In Russ).
doi - Chaulin AM, Duplyakov DV. Environmental factors and cardiovascular diseases. Hygiene and Sanitation. 2021;100(3):223-228. (in Russian).
doi - WHO-World Health Organization. 2018. WHO ambient (outdoor) air quality database summary results, update 2018. URL: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/ambient-air-pollution (Accessed August 1, 2021).
- Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389(10082):1907-1918.
doi - Du Y, Xu X, Chu M, Guo Y, Wang J. Air particulate matter and cardiovascular disease: the epidemiological, biomedical and clinical evidence. J Thorac Dis. 2016;8(1):E8-E19.
- Chaulin AM, Duplyakov DV. Comorbidity in chronic obstructive pulmonary disease and cardiovascular disease. Cardiovascular Therapy and Prevention. 2021;20(3):2539. (in Russian).
doi - Chaulin AM. Cardiac troponins: current information on the main analytical characteristics of determination methods and new diagnostic possibilities. Medwave. 2021;21(11):e8498.
doi pubmed - Vriens A, Nawrot TS, Janssen BG, Baeyens W, Bruckers L, Covaci A, De Craemer S, et al. Exposure to Environmental Pollutants and Their Association with Biomarkers of Aging: A Multipollutant Approach. Environ Sci Technol. 2019;53(10):5966-5976.
doi pubmed - Gan WQ, Allen RW, Brauer M, Davies HW, Mancini GB, Lear SA. Long-term exposure to traffic-related air pollution and progression of carotid artery atherosclerosis: a prospective cohort study. BMJ Open. 2014;4(4):e004743.
doi pubmed - Tian Y, Liu H, Wu Y, Si Y, Song J, Cao Y, Li M, et al. Association between ambient fine particulate pollution and hospital admissions for cause specific cardiovascular disease: time series study in 184 major Chinese cities. BMJ. 2019;367:l6572.
doi pubmed - Xu Q, Wang S, Guo Y, Wang C, Huang F, Li X, Gao Q, et al. Acute exposure to fine particulate matter and cardiovascular hospital emergency room visits in Beijing, China. Environ Pollut. 2017;220(Pt A):317-327.
doi pubmed - Xu H, Brook RD, Wang T, Song X, Feng B, Yi T, Liu S, et al. Short-term effects of ambient air pollution and outdoor temperature on biomarkers of myocardial damage, inflammation and oxidative stress in healthy adults. Environ Epidemiol. 2019;3(6):e078.
doi pubmed - Zhang S, Breitner S, Cascio WE, Devlin RB, Neas LM, Ward-Caviness C, Diaz-Sanchez D, et al. Association between short-term exposure to ambient fine particulate matter and myocardial injury in the CATHGEN cohort. Environ Pollut. 2021;275:116663.
doi pubmed - Huang CH, Lin LY, Tsai MS, Hsu CY, Chen HW, Wang TD, Chang WT, et al. Acute cardiac dysfunction after short-term diesel exhaust particles exposure. Toxicol Lett. 2010;192(3):349-355.
doi pubmed - Vieira JL, Guimaraes GV, de Andre PA, Cruz FD, Saldiva PH, Bocchi EA. Respiratory Filter Reduces the Cardiovascular Effects Associated With Diesel Exhaust Exposure: A Randomized, Prospective, Double-Blind, Controlled Study of Heart Failure: The FILTER-HF Trial. JACC Heart Fail. 2016;4(1):55-64.
doi pubmed - Eze UU, Eke IG, Anakwue RC, Oguejiofor CF, Onyejekwe OB, Udeani IJ, Onunze CJ, et al. Effects of Controlled Generator Fume Emissions on the Levels of Troponin I, C-Reactive Protein and Oxidative Stress Markers in Dogs: Exploring Air Pollution-Induced Cardiovascular Disease in a Low-Resource Country. Cardiovasc Toxicol. 2021;21(12):1019-1032.
doi pubmed - Wei Z, Wang L, Chen M, Zheng Y. The 2013 severe haze over the southern HeBei, China: PM2.5, composition and source apportionment. Atmospheric Pollution Research. 2014;5(4):759-768.
doi - Werner M, Kryza M, Dore AJ. Differences in the spatial distribution and chemical composition of PM10 between the UK and Poland. Environ Model Assess. 2014;19:179-192.
doi - Philip G, Nayak AS, Berger WE, Leynadier F, Vrijens F, Dass SB, Reiss TF. The effect of montelukast on rhinitis symptoms in patients with asthma and seasonal allergic rhinitis. Curr Med Res Opin. 2004;20(10):1549-1558.
doi pubmed - Wang G, Zheng X, Duan H, Dai Y, Niu Y, Gao J, Chang Z, et al. High-content analysis of particulate matters-induced oxidative stress and organelle dysfunction in vitro. Toxicol In Vitro. 2019;59:263-274.
doi pubmed - Zeka A, Sullivan JR, Vokonas PS, Sparrow D, Schwartz J. Inflammatory markers and particulate air pollution: characterizing the pathway to disease. Int J Epidemiol. 2006;35(5):1347-1354.
doi pubmed - Meng X, Zhang Y, Yang KQ, Yang YK, Zhou XL. Potential Harmful Effects of PM2.5 on Occurrence and Progression of Acute Coronary Syndrome: Epidemiology, Mechanisms, and Prevention Measures. Int J Environ Res Public Health. 2016;13(8):748.
doi pubmed - Kim SY, Sheppard L, Kaufman JD, Bergen S, Szpiro AA, Larson TV, Adar SD, et al. Individual-level concentrations of fine particulate matter chemical components and subclinical atherosclerosis: a cross-sectional analysis based on 2 advanced exposure prediction models in the multi-ethnic study of atherosclerosis. Am J Epidemiol. 2014;180(7):718-728.
doi pubmed - Sun M, Kaufman JD, Kim SY, Larson TV, Gould TR, Polak JF, Budoff MJ, et al. Particulate matter components and subclinical atherosclerosis: common approaches to estimating exposure in a Multi-Ethnic Study of Atherosclerosis cross-sectional study. Environ Health. 2013;12:39.
doi pubmed - Su R, Jin X, Li H, Huang L, Li Z. The mechanisms of PM2.5 and its main components penetrate into HUVEC cells and effects on cell organelles. Chemosphere. 2020;241:125127.
doi pubmed - Miao X, Li W, Niu B, Li J, Sun J, Qin M, Zhou Z. Mitochondrial dysfunction in endothelial cells induced by airborne fine particulate matter (<2.5 mum). J Appl Toxicol. 2019;39(10):1424-1432.
doi pubmed - Wang Y, Zhang M, Li Z, Yue J, Xu M, Zhang Y, Yung KKL, et al. Fine particulate matter induces mitochondrial dysfunction and oxidative stress in human SH-SY5Y cells. Chemosphere. 2019;218:577-588.
doi pubmed - Li R, Kou X, Geng H, Xie J, Yang Z, Zhang Y, Cai Z, et al. Effect of ambient PM(2.5) on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol. 2015;28(3):408-418.
doi pubmed - Kunzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, Thomas D, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113(2):201-206.
doi pubmed - Breton CV, Wang X, Mack WJ, Berhane K, Lopez M, Islam TS, Feng M, et al. Childhood air pollutant exposure and carotid artery intima-media thickness in young adults. Circulation. 2012;126(13):1614-1620.
doi pubmed - Bauer M, Moebus S, Mohlenkamp S, Dragano N, Nonnemacher M, Fuchsluger M, Kessler C, et al. Urban particulate matter air pollution is associated with subclinical atherosclerosis: results from the HNR (Heinz Nixdorf Recall) study. J Am Coll Cardiol. 2010;56(22):1803-1808.
doi pubmed - Adar SD, Sheppard L, Vedal S, Polak JF, Sampson PD, Diez Roux AV, Budoff M, et al. Fine particulate air pollution and the progression of carotid intima-medial thickness: a prospective cohort study from the multi-ethnic study of atherosclerosis and air pollution. PLoS Med. 2013;10(4):e1001430.
doi pubmed - Diez Roux AV, Auchincloss AH, Franklin TG, Raghunathan T, Barr RG, Kaufman J, Astor B, et al. Long-term exposure to ambient particulate matter and prevalence of subclinical atherosclerosis in the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol. 2008;167(6):667-675.
doi pubmed - Hoffmann B, Moebus S, Mohlenkamp S, Stang A, Lehmann N, Dragano N, Schmermund A, et al. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation. 2007;116(5):489-496.
doi pubmed - Kaufman JD, Adar SD, Barr RG, Budoff M, Burke GL, Curl CL, Daviglus ML, et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): a longitudinal cohort study. Lancet. 2016;388(10045):696-704.
doi - Hoffmann B, Moebus S, Kroger K, Stang A, Mohlenkamp S, Dragano N, Schmermund A, et al. Residential exposure to urban air pollution, ankle-brachial index, and peripheral arterial disease. Epidemiology. 2009;20(2):280-288.
doi pubmed - Kalsch H, Hennig F, Moebus S, Mohlenkamp S, Dragano N, Jakobs H, Memmesheimer M, et al. Are air pollution and traffic noise independently associated with atherosclerosis: the Heinz Nixdorf Recall Study. Eur Heart J. 2014;35(13):853-860.
doi pubmed - Brook RD, Rajagopalan S, Pope CA, 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331-2378.
doi pubmed - Crouse DL, Peters PA, van Donkelaar A, Goldberg MS, Villeneuve PJ, Brion O, Khan S, et al. Risk of nonaccidental and cardiovascular mortality in relation to long-term exposure to low concentrations of fine particulate matter: a Canadian national-level cohort study. Environ Health Perspect. 2012;120(5):708-714.
doi pubmed - Pinault L, Tjepkema M, Crouse DL, Weichenthal S, van Donkelaar A, Martin RV, Brauer M, et al. Risk estimates of mortality attributed to low concentrations of ambient fine particulate matter in the Canadian community health survey cohort. Environ Health. 2016;15:18.
doi pubmed - Christidis T, Erickson AC, Pappin AJ, Crouse DL, Pinault LL, Weichenthal SA, Brook JR, et al. Low concentrations of fine particle air pollution and mortality in the Canadian Community Health Survey cohort. Environ Health. 2019;18(1):84.
doi pubmed - Huynh Q, Marwick TH, Venkataraman P, Knibbs LD, Johnston FH, Negishi K. Long-term exposure to ambient air pollution is associated with coronary artery calcification among asymptomatic adults. Eur Heart J Cardiovasc Imaging. 2021;22(8):922-929.
doi pubmed - Lin J, Zheng H, Xia P, Cheng X, Wu W, Li Y, Ma C, et al. Long-term ambient PM2.5 exposure associated with cardiovascular risk factors in Chinese less educated population. BMC Public Health. 2021;21(1):2241.
doi pubmed - Ren Z, Liu X, Liu T, Chen D, Jiao K, Wang X, Suo J, et al. Effect of ambient fine particulates (PM2.5) on hospital admissions for respiratory and cardiovascular diseases in Wuhan, China. Respir Res. 2021;22(1):128.
doi pubmed - Liang R, Chen R, Yin P, van Donkelaar A, Martin RV, Burnett R, Cohen AJ, et al. Associations of long-term exposure to fine particulate matter and its constituents with cardiovascular mortality: A prospective cohort study in China. Environ Int. 2022;162:107156.
doi pubmed - Huang W, Wang G, Lu SE, Kipen H, Wang Y, Hu M, Lin W, et al. Inflammatory and oxidative stress responses of healthy young adults to changes in air quality during the Beijing Olympics. Am J Respir Crit Care Med. 2012;186(11):1150-1159.
doi pubmed - Rich DQ, Kipen HM, Huang W, Wang G, Wang Y, Zhu P, Ohman-Strickland P, et al. Association between changes in air pollution levels during the Beijing Olympics and biomarkers of inflammation and thrombosis in healthy young adults. JAMA. 2012;307(19):2068-2078.
doi pubmed - Roberts JW, Wallace LA, Camann DE, Dickey P, Gilbert SG, Lewis RG, Takaro TK. Monitoring and reducing exposure of infants to pollutants in house dust. Rev Environ Contam Toxicol. 2009;201:1-39.
doi pubmed - Barn P, Gombojav E, Ochir C, Laagan B, Beejin B, Naidan G, Boldbaatar B, et al. The effect of portable HEPA filter air cleaners on indoor PM2.5 concentrations and second hand tobacco smoke exposure among pregnant women in Ulaanbaatar, Mongolia: The UGAAR randomized controlled trial. Sci Total Environ. 2018;615:1379-1389.
doi pubmed - Maestas MM, Brook RD, Ziemba RA, Li F, Crane RC, Klaver ZM, Bard RL, et al. Reduction of personal PM2.5 exposure via indoor air filtration systems in Detroit: an intervention study. J Expo Sci Environ Epidemiol. 2019;29(4):484-490.
doi pubmed - Zhu Y, Song X, Wu R, Fang J, Liu L, Wang T, Liu S, et al. A review on reducing indoor particulate matter concentrations from personal-level air filtration intervention under real-world exposure situations. Indoor Air. 2021;31(6):1707-1721.
doi pubmed - Liang S, Zhao T, Xu Q, Duan J, Sun Z. Evaluation of fine particulate matter on vascular endothelial function in vivo and in vitro. Ecotoxicol Environ Saf. 2021;222:112485.
doi pubmed - Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19(11):2648-2658.
doi pubmed - Mannucci PM, Harari S, Franchini M. Novel evidence for a greater burden of ambient air pollution on cardiovascular disease. Haematologica. 2019;104(12):2349-2357.
doi pubmed - Back M, Yurdagul A, Jr., Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16(7):389-406.
doi pubmed - Chaulin AM, Grigoryeva YuV, Duplyakov DV. About the role of immuno-inflammatory mechanisms in the pathogenesis of atherosclerosis. European Journal of Natural History. 2020;5:2-6.
doi - Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4(11):e002270.
doi pubmed - Brook RD, Urch B, Dvonch JT, Bard RL, Speck M, Keeler G, Morishita M, et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54(3):659-667.
doi pubmed - Krishnan RM, Adar SD, Szpiro AA, Jorgensen NW, Van Hee VC, Barr RG, O'Neill MS, et al. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution). J Am Coll Cardiol. 2012;60(21):2158-2166.
doi pubmed - Pope CA, 3rd, Bhatnagar A, McCracken JP, Abplanalp W, Conklin DJ, O'Toole T. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circ Res. 2016;119(11):1204-1214.
doi pubmed - Wang T, Chiang ET, Moreno-Vinasco L, Lang GD, Pendyala S, Samet JM, Geyh AS, et al. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol. 2010;42(4):442-449.
doi pubmed - Wang M, Hou ZH, Xu H, Liu Y, Budoff MJ, Szpiro AA, Kaufman JD, et al. Association of Estimated Long-term Exposure to Air Pollution and Traffic Proximity With a Marker for Coronary Atherosclerosis in a Nationwide Study in China. JAMA Netw Open. 2019;2(6):e196553.
doi pubmed - Zhou Z, Shao T, Qin M, Miao X, Chang Y, Sheng W, Wu F, et al. The effects of autophagy on vascular endothelial cells induced by airborne PM2.5. J Environ Sci (China). 2018;66:182-187.
doi pubmed - Montiel-Davalos A, Alfaro-Moreno E, Lopez-Marure R. PM2.5 and PM10 induce the expression of adhesion molecules and the adhesion of monocytic cells to human umbilical vein endothelial cells. Inhal Toxicol. 2007;19(Suppl 1):91-98.
doi pubmed - Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105(8):1049-1056.
doi pubmed - Geng J, Liu H, Ge P, Hu T, Zhang Y, Zhang X, Xu B, et al. PM2.5 promotes plaque vulnerability at different stages of atherosclerosis and the formation of foam cells via TLR4/MyD88/NFkappaB pathway. Ecotoxicol Environ Saf. 2019;176:76-84.
doi pubmed - Mollace V, Gliozzi M, Musolino V, Carresi C, Muscoli S, Mollace R, Tavernese A, et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int J Cardiol. 2015;184:152-158.
doi pubmed - Konstantinov IE, Mejevoi N, Anichkov NM. Nikolai N. Anichkov and his theory of atherosclerosis. Tex Heart Inst J. 2006;33(4):417-423.
- Slawsky E, Ward-Caviness CK, Neas L, Devlin RB, Cascio WE, Russell AG, Huang R, et al. Evaluation of PM2.5 air pollution sources and cardiovascular health. Environ Epidemiol. 2021;5(3):e157.
doi pubmed - Valdez RB, Al-Hamdan MZ, Tabatabai M, Hood DB, Im W, Wilus D, Nori-Sarma A, et al. Association of cardiovascular disease and long-term exposure to fine particulate matter (PM2.5) in the Southeastern United States. Atmosphere. 2021;12(8):947.
doi - Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, et al. Particulate matter exposure and stress hormone levels: a randomized, double-blind, crossover trial of air purification. Circulation. 2017;136(7):618-627.
doi pubmed - Guan L, Geng X, Shen J, Yip J, Li F, Du H, Ji Z, et al. PM2.5 inhalation induces intracranial atherosclerosis which may be ameliorated by omega 3 fatty acids. Oncotarget. 2018;9(3):3765-3778.
doi pubmed - Mao S, Chen G, Liu F, Li N, Wang C, Liu Y, Liu S, et al. Long-term effects of ambient air pollutants to blood lipids and dyslipidemias in a Chinese rural population. Environ Pollut. 2020;256:113403.
doi pubmed - McGuinn LA, Schneider A, McGarrah RW, Ward-Caviness C, Neas LM, Di Q, Schwartz J, et al. Association of long-term PM2.5 exposure with traditional and novel lipid measures related to cardiovascular disease risk. Environ Int. 2019;122:193-200.
doi pubmed - Bell G, Mora S, Greenland P, Tsai M, Gill E, Kaufman JD. Association of air pollution exposures with high-density lipoprotein cholesterol and particle number: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37(5):976-982.
doi pubmed - Holme SAN, Sigsgaard T, Holme JA, Holst GJ. Effects of particulate matter on atherosclerosis: a link via high-density lipoprotein (HDL) functionality? Part Fibre Toxicol. 2020;17(1):36.
doi pubmed - Ramanathan G, Yin F, Speck M, Tseng CH, Brook JR, Silverman F, Urch B, et al. Effects of urban fine particulate matter and ozone on HDL functionality. Part Fibre Toxicol. 2016;13(1):26.
doi pubmed - Freeman SR, Jin X, Anzinger JJ, Xu Q, Purushothaman S, Fessler MB, Addadi L, et al. ABCG1-mediated generation of extracellular cholesterol microdomains. J Lipid Res. 2014;55(1):115-127.
doi pubmed - Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, Perisa D, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127(8):891-904.
doi pubmed - Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294(23):3003-3010.
doi pubmed - Du X, Jiang S, Zeng X, Zhang J, Pan K, Zhou J, Xie Y, et al. Air pollution is associated with the development of atherosclerosis via the cooperation of CD36 and NLRP3 inflammasome in ApoE(-/-) mice. Toxicol Lett. 2018;290:123-132.
doi pubmed - Rao X, Zhong J, Maiseyeu A, Gopalakrishnan B, Villamena FA, Chen LC, Harkema JR, et al. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res. 2014;115(9):770-780.
doi pubmed - Shi Y, Hu J, Geng J, Hu T, Wang B, Yan W, Jiang Y, et al. Berberine treatment reduces atherosclerosis by mediating gut microbiota in apoE-/- mice. Biomed Pharmacother. 2018;107:1556-1563.
doi pubmed - Crobeddu B, Aragao-Santiago L, Bui LC, Boland S, Baeza Squiban A. Oxidative potential of particulate matter 2.5 as predictive indicator of cellular stress. Environ Pollut. 2017;230:125-133.
doi pubmed - Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13(6):368-380.
doi pubmed - Sotty J, Kluza J, De Sousa C, Tardivel M, Antherieu S, Alleman LY, Canivet L, et al. Mitochondrial alterations triggered by repeated exposure to fine (PM2.5-0.18) and quasi-ultrafine (PM0.18) fractions of ambient particulate matter. Environ Int. 2020;142:105830.
doi pubmed - Liu J, Liang S, Du Z, Zhang J, Sun B, Zhao T, Yang X, et al. PM2.5 aggravates the lipid accumulation, mitochondrial damage and apoptosis in macrophage foam cells. Environ Pollut. 2019;249:482-490.
doi pubmed - Tian G, Wang J, Lu Z, Wang H, Zhang W, Ding W, Zhang F. Indirect effect of PM1 on endothelial cells via inducing the release of respiratory inflammatory cytokines. Toxicol In Vitro. 2019;57:203-210.
doi pubmed - Prueitt RL, Cohen JM, Goodman JE. Evaluation of atherosclerosis as a potential mode of action for cardiovascular effects of particulate matter. Regul Toxicol Pharmacol. 2015;73(2 Suppl):S1-15.
doi pubmed - Hassan L, Pecht T, Goldstein N, Haim Y, Kloog I, Yarza S, Sarov B, et al. The effects of ambient particulate matter on human adipose tissue. J Toxicol Environ Health A. 2019;82(9):564-576.
doi pubmed - Bai Y, Sun Q. Fine particulate matter air pollution and atherosclerosis: Mechanistic insights. Biochim Biophys Acta. 2016;1860(12):2863-2868.
doi pubmed - Wan Q, Ding T, Xu Y, Zheng C, Tu M, Zhao T. Urban fine particulate air pollution exposure promotes atherosclerosis in apolipoprotein E-deficient mice by activating perivascular adipose tissue inflammation via the Wnt5a/Ror2 signaling pathway. Ecotoxicol Environ Saf. 2021;227:112912.
doi pubmed - Liu C, Bai Y, Xu X, Sun L, Wang A, Wang TY, Maurya SK, et al. Exaggerated effects of particulate matter air pollution in genetic type II diabetes mellitus. Part Fibre Toxicol. 2014;11:27.
doi pubmed - Yoo HJ, Choi KM. Adipokines as a novel link between obesity and atherosclerosis. World J Diabetes. 2014;5(3):357-363.
doi pubmed - Nakamura K, Fuster JJ, Walsh K. Adipokines: a link between obesity and cardiovascular disease. J Cardiol. 2014;63(4):250-259.
doi pubmed - Raman P, Khanal S. Leptin in atherosclerosis: focus on macrophages, endothelial and smooth muscle cells. Int J Mol Sci. 2021;22(11):5446.
doi pubmed - Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne). 2013;4:71.
doi pubmed - Chaulin A. Cardiac troponins: contemporary biological data and new methods of determination. Vasc Health Risk Manag. 2021;17:299-316.
doi pubmed - Chaulin AM, Duplyakova PD, Duplyakov DV. Circadian rhythms of cardiac troponins: mechanisms and clinical significance. Russian Journal of Cardiology. 2020;25(3S):4061.
doi - Kim KN, Kim JH, Jung K, Hong YC. Associations of air pollution exposure with blood pressure and heart rate variability are modified by oxidative stress genes: A repeated-measures panel among elderly urban residents. Environ Health. 2016;15:47.
doi pubmed - Pieters N, Plusquin M, Cox B, Kicinski M, Vangronsveld J, Nawrot TS. An epidemiological appraisal of the association between heart rate variability and particulate air pollution: a meta-analysis. Heart. 2012;98(15):1127-1135.
doi pubmed - Lee MS, Eum KD, Fang SC, Rodrigues EG, Modest GA, Christiani DC. Oxidative stress and systemic inflammation as modifiers of cardiac autonomic responses to particulate air pollution. Int J Cardiol. 2014;176(1):166-170.
doi pubmed - Pei Y, Jiang R, Zou Y, Wang Y, Zhang S, Wang G, Zhao J, et al. Effects of fine particulate matter (PM2.5) on systemic oxidative stress and cardiac function in ApoE(-/-) mice. Int J Environ Res Public Health. 2016;13(5):484.
doi pubmed - Huang F, Wang P, Pan X, Wang Y, Ren S. Effects of short-term exposure to particulate matters on heart rate variability: A systematic review and meta-analysis based on controlled animal studies. Environ Pollut. 2020;256:113306.
doi pubmed - Fuks KB, Weinmayr G, Hennig F, Tzivian L, Moebus S, Jakobs H, Memmesheimer M, et al. Association of long-term exposure to local industry- and traffic-specific particulate matter with arterial blood pressure and incident hypertension. Int J Hyg Environ Health. 2016;219(6):527-535.
doi pubmed - Auchincloss AH, Diez Roux AV, Dvonch JT, Brown PL, Barr RG, Daviglus ML, Goff DC, et al. Associations between recent exposure to ambient fine particulate matter and blood pressure in the Multi-ethnic Study of Atherosclerosis (MESA). Environ Health Perspect. 2008;116(4):486-491.
doi pubmed - Ying Z, Xu X, Bai Y, Zhong J, Chen M, Liang Y, Zhao J, et al. Long-term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: a role for hypothalamic inflammation. Environ Health Perspect. 2014;122(1):79-86.
doi pubmed - Tobaldini E, Bollati V, Prado M, Fiorelli EM, Pecis M, Bissolotti G, Albetti B, et al. Acute particulate matter affects cardiovascular autonomic modulation and IFN-gamma methylation in healthy volunteers. Environ Res. 2018;161:97-103.
doi pubmed - Chaulin AM. Diagnostic value of highly sensitive cardiac troponins and mechanisms of their increase in serum and urine in arterial hypertension. La Rivista Italiana della Medicina di Laboratorio. 2021;17(2):99-107.
doi pubmed - Chaulin A, Duplyakov D. Cardiac troponins: current data on the diagnostic value and analytical characteristics of new determination methods. Cor Vasa. 2021;63:486-493.
doi - Gerhard GT, Duell PB. Homocysteine and atherosclerosis. Curr Opin Lipidol. 1999;10(5):417-428.
doi pubmed - Chaulin A. Clinical and diagnostic value of highly sensitive cardiac troponins in arterial hypertension. Vasc Health Risk Manag. 2021;17:431-443.
doi pubmed - Hurtubise J, McLellan K, Durr K, Onasanya O, Nwabuko D, Ndisang JF. The different facets of dyslipidemia and hypertension in atherosclerosis. Curr Atheroscler Rep. 2016;18(12):82.
doi pubmed - Ivanovic B, Tadic M. Hypercholesterolemia and hypertension: two sides of the same coin. Am J Cardiovasc Drugs. 2015;15(6):403-414.
doi pubmed - Burke AP, Farb A, Malcom GT, Liang Y, Smialek JE, Virmani R. Plaque rupture and sudden death related to exertion in men with coronary artery disease. JAMA. 1999;281(10):921-926.
doi pubmed - Jin SX, Shen LH, Nie P, Yuan W, Hu LH, Li DD, Chen XJ, et al. Endogenous renovascular hypertension combined with low shear stress induces plaque rupture in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32(10):2372-2379.
doi pubmed - Sierra C, Coca A, Schiffrin EL. Vascular mechanisms in the pathogenesis of stroke. Curr Hypertens Rep. 2011;13(3):200-207.
doi pubmed - Nemmar A, Hoet PH, Dinsdale D, Vermylen J, Hoylaerts MF, Nemery B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation. 2003;107(8):1202-1208.
doi pubmed - Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest. 2007;117(10):2952-2961.
doi pubmed - Ruckerl R, Phipps RP, Schneider A, Frampton M, Cyrys J, Oberdorster G, Wichmann HE, et al. Ultrafine particles and platelet activation in patients with coronary heart disease—results from a prospective panel study. Part Fibre Toxicol. 2007;4:1.
doi pubmed - Baccarelli A, Zanobetti A, Martinelli I, Grillo P, Hou L, Giacomini S, Bonzini M, et al. Effects of exposure to air pollution on blood coagulation. J Thromb Haemost. 2007;5(2):252-260.
doi pubmed - Tang L, Shi S, Wang B, Liu L, Yang Y, Sun X, Ni Z, et al. Effect of urban air pollution on CRP and coagulation: a study on inpatients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med. 2021;21(1):296.
doi pubmed - Chaulin AM, Duplyakov DV. On the potential effect of circadian rhythms of cardiac troponins on the diagnosis of acute myocardial infarction. Signa Vitae. 2021;17(3):79-84.
doi - Chaulin AM, Dupliakov DV. Physical activity and cardiac markers: part 1. Human Sport Medicine. 2022;22(2):15-28. (In Russ).
doi - Muscente F, De Caterina R. New insights from the MESA study: increased high-sensitivity troponins as a cardiovascular risk factor. Eur Heart J Suppl. 2021;23(Suppl E):E68-E72.
doi pubmed - Chauin A. The main causes and mechanisms of increase in cardiac troponin concentrations other than acute myocardial infarction (Part 1): physical exertion, inflammatory heart disease, pulmonary embolism, renal failure, sepsis. Vasc Health Risk Manag. 2021;17:601-617.
doi pubmed - Chaulin AM. Elevation mechanisms and diagnostic consideration of cardiac troponins under conditions not associated with myocardial infarction. Part 2. Life (Basel). 2021;11(11):1175.
doi pubmed - Chaulin AM, Abashina OE, Duplyakov DV. High-sensitivity cardiac troponins: detection and central analytical characteristics. Cardiovascular Therapy and Prevention. 2021;20(2):2590. (In Russ).
doi - Huang YC, Ghio AJ, Stonehuerner J, McGee J, Carter JD, Grambow SC, Devlin RB. The role of soluble components in ambient fine particles-induced changes in human lungs and blood. Inhal Toxicol. 2003;15(4):327-342.
doi pubmed - Ghio AJ, Carraway MS, Madden MC. Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. J Toxicol Environ Health B Crit Rev. 2012;15(1):1-21.
doi pubmed - Ye D, Klein M, Mulholland JA, Russell AG, Weber R, Edgerton ES, Chang HH, et al. Estimating acute cardiovascular effects of ambient PM2.5 metals. Environ Health Perspect. 2018;126(2):027007.
doi pubmed - Mills NL, Miller MR, Lucking AJ, Beveridge J, Flint L, Boere AJ, Fokkens PH, et al. Combustion-derived nanoparticulate induces the adverse vascular effects of diesel exhaust inhalation. Eur Heart J. 2011;32(21):2660-2671.
doi pubmed - Lozano-Sabido ED, Berrios-Barcenas EA, Cazares-Diazleal AC, Viveros-Renteria E, Alvarez-Mosquera JB, Portos-Silva JM, Kiamco-Castillo CR. "ST-elevation myocardial infarction associated with air pollution levels in Mexico City". Int J Cardiol Heart Vasc. 2021;35:100846.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.