











| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website http://www.cardiologyres.org |
Case Report
Volume 8, Number 2, April 2017, pages 57-62
Lupus-Negative Libman-Sacks Endocarditis Complicated by Catastrophic Antiphospholipid Syndrome
Ghulam Murtazaa, b, Joy Iskandara, Tara Humphreya, Sujeen Adhikaria, Aneesh Kuruvillaa
aDepartment of Internal Medicine, Advocate Christ Medical Center, Oak Lawn, IL, USA
bCorresponding Author: Ghulam Murtaza, Department of Internal Medicine, Advocate Christ Medical Center, 4440 W 95th St, Oak Lawn, IL 60453, USA
Manuscript accepted for publication April 14, 2017
Short title: Lupus-Negative Libman-Sacks Endocarditis
doi: https://doi.org/10.14740/cr534e
| Abstract | ▴Top |
Libman-Sacks endocarditis is characterized by sterile and verrucous lesions that predominantly affect the aortic and mitral valves. In most cases, patients do not have significant valvular dysfunction. However, patients with significant valvular dysfunction may present with serious complications such as cardiac failure, arrhythmias, and thromboembolic events. Recently, association of Libman-Sacks endocarditis with antiphospholipid antibody syndrome (APS) has been made. APS is most commonly defined by venous and arterial thrombosis, recurrent pregnancy loss, and thrombocytopenia. While the syndrome can be a primary syndrome, it is usually secondary to systemic lupus erythematosus. Catastrophic antiphospholipid syndrome (CAPS) can be a life-threatening presentation of APS and can occur in 1% of patients with antiphospholipid syndrome. We present a very rare case of a young female patient with lupus-negative Libman-Sacks endocarditis complicated by CAPS.
Keywords: Libman-Sacks endocarditis; Antiphospholipid antibody syndrome; Catastrophic antiphospholipid syndrome; Cardiac failure
| Introduction | ▴Top |
Libman-Sacks endocarditis was first described in patients with autoimmune disease and systemic lupus erythematosus (SLE). Libman-Sacks endocarditis is characterized by sterile and verrucous lesions that mostly affect the mitral and aortic valves. In most cases, patients do not have significant valvular dysfunction. However, patients with significant valvular dysfunction may present with serious complications such as cardiac failure, arrhythmias, and thromboembolic events [1, 2]. Recently, association of Libman-Sacks endocarditis with antiphospholipid antibody syndrome (APS) has been made [2]. APS is most commonly defined by venous and arterial thrombosis, recurrent pregnancy loss, and thrombocytopenia. While the syndrome can be a primary syndrome, it is usually secondary to SLE. Catastrophic antiphospholipid syndrome (CAPS) can be a life-threatening presentation of APS and can occur in 1% of patients with APS [3]. We present a rare case of a young female patient with lupus-negative Libman-Sacks endocarditis complicated by CAPS.
| Case Report | ▴Top |
A 22-year-old Caucasian female with no past medical history presented to the emergency department after being found by her boyfriend unconscious at home. The patient’s last known normal was 11 h prior. The mother reported that the patient had complaints of generalized malaise and fatigue over the last 2 months, but otherwise she was healthy. No sick contacts were reported. Family history was significant for Hashimoto’s thyroiditis in the mother and celiac disease in the father. Patient had a two pack year history of smoking and had recently quit 3 months ago. The only medication patient was taking was an oral contraceptive. She was an occasional alcohol and marijuana user but did not use any other recreational drugs.
On presentation to the emergency department, blood pressure was 90/61 mm Hg, heart rate was 127 beats per minute, temperature was 38.9 °C, respiratory rate was 28 breaths per minute and saturation was 87% on room air. The patient was subsequently intubated for airway protection. On physical exam, pupils were equal and reactive, 2/6 holosystolic murmur was heard on the left lower sternal border, and pulses were diminished throughout. Extremities revealed cold, dusky fingers with mottling of toes bilaterally. On neurological exam, there were no spontaneous movements of left upper extremity and minimal movements of the left lower extremity.
Labs on admission are listed in Table 1.
 Click to view | Table 1. Laboratory Tests on Admission |
The 2D transthoracic echocardiogram showed an ejection fraction of 15% and severe mitral regurgitation. Given the altered mental status, CT of head was obtained which showed extensive multifocal areas of hypoattenuation throughout the bilateral frontal, parietal, occipital and right temporal lobes consistent with multifocal infarction concerning for cardioembolic etiology (Fig. 1).
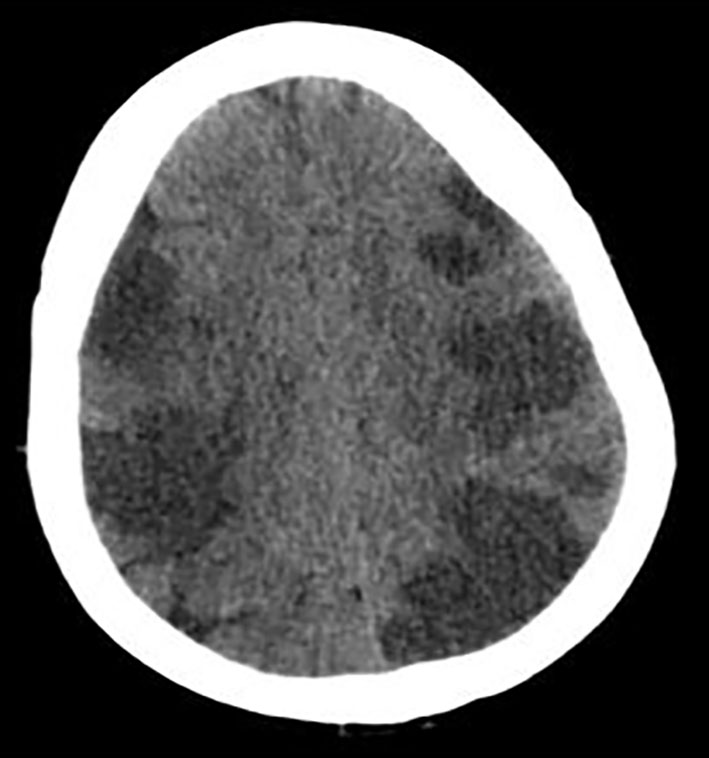 Click for large image | Figure 1. CT of head without contrast showing extensive multifocal areas of hypoattentuation throughout the bilateral frontal, parietal, occipital, and right > left temporal lobes. No mass effect or midline shift or hemorrhage was seen. |
Given the fever, leukocytosis and suspected cardioembolic phenomenon causing infarctions in the brain, there was concern for bacterial endocarditis. Hence, transesophageal echocardiogram (TEE) was performed which showed a 1 cm vegetation at the anterior mitral leaflet (Fig. 2, Supplementary videos 1 and 2, www.cardiologyres.org). The patient was started on broad spectrum antibiotics, vancomycin, and ceftriaxone for presumed bacterial endocarditis with systemic emboli. However, blood cultures obtained from two different sites (peripheral and antecubital) prior to administration of antibiotics remained negative throughout the hospital stay.
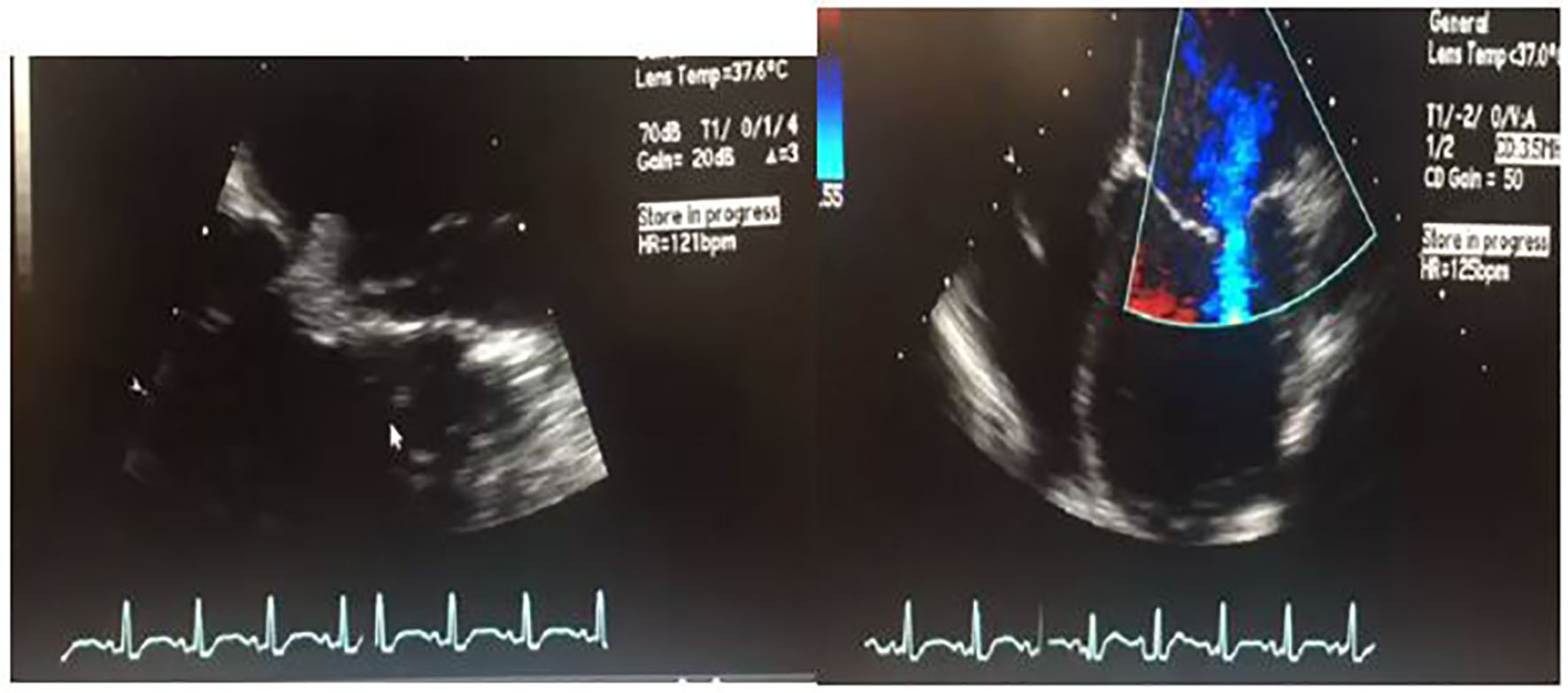 Click for large image | Figure 2. 2D transesophageal ultrasound. Image on the left shows a thickened mitral valve with a 1 cm vegetation that can be seen on the anterior mitral leaflet. Image on the right is a four-chamber color flow Doppler view showing biventricular dilatation, severe left ventricular dysfunction. For orientation purposes, left ventricle is the bottom right chamber. Video of both views is attached as a supplementary file. |
Autoimmune workup was negative. Hematological workup showed an isolated lupus anticoagulant with normal anti-cardiolipin antibodies. Repeat lupus anticoagulant a few days later was negative. Patient likely had disseminated intravascular coagulation that was resolving. Thus, hypercoagulable workup was not performed with presumption of false positive findings in the setting of critical illness along with a vegetation that was showering emboli.
On day 8, metabolic laboratory abnormalities improved but patient’s neurological status worsened and she no longer opened her eyes spontaneously. That she had multifocal infarctions based on her initial head CT scan was already known. Although her initial head CT had shown multiple infarctions, her abrupt neurological decline was unclear. This prompted us to perform an MRI of the brain. It showed increased signal on DWI throughout bilateral frontal, parietal, and occipital lobes consistent with new/ongoing acute infarcts (Fig. 3). After discussion with family and consultants, we decided to start her on therapeutic anticoagulation with heparin.
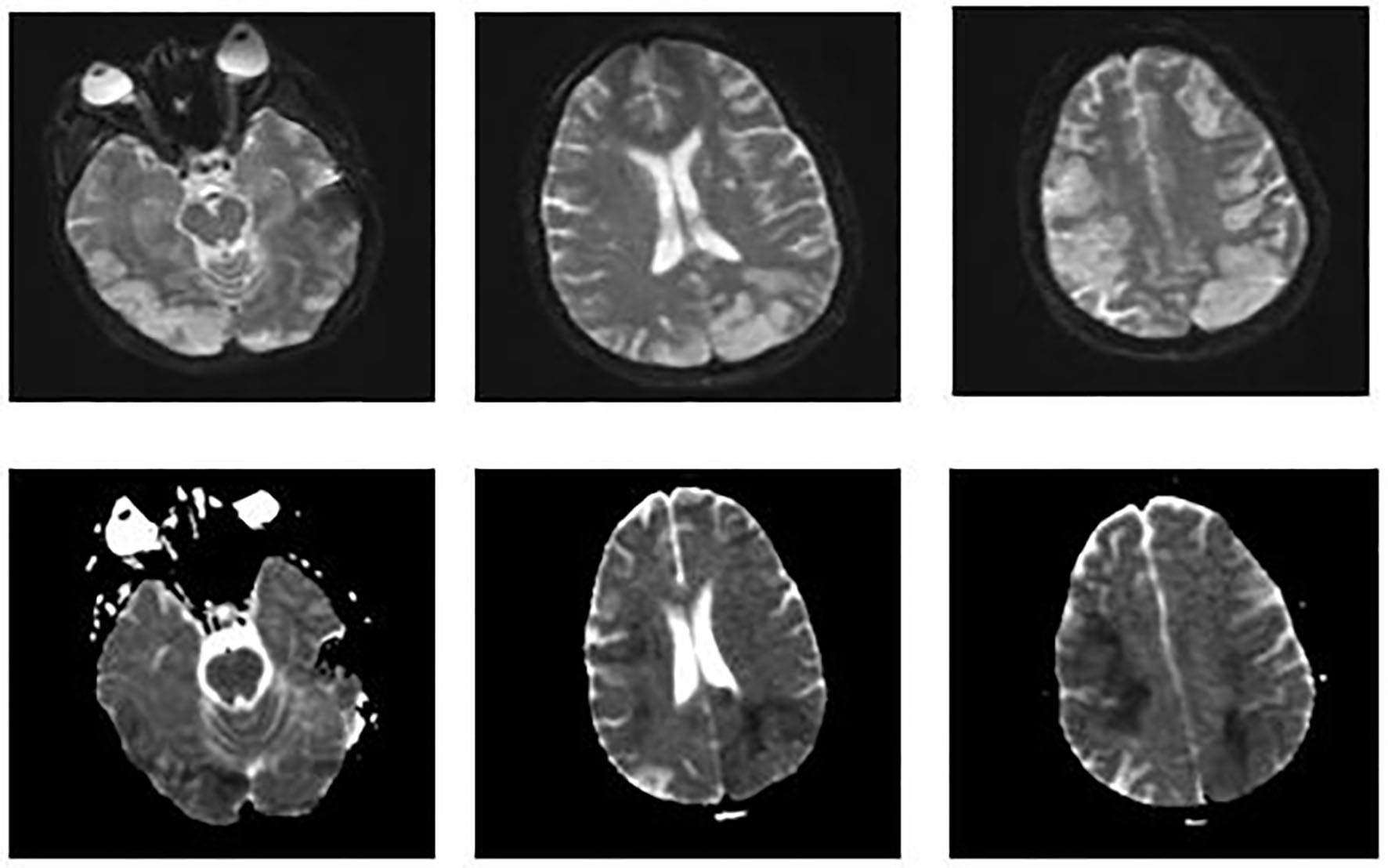 Click for large image | Figure 3. MRI of the brain. Images on the top show increased signal on diffusion weighted imaging (DWI) throughout the bilateral frontal, parietal, and occipital lobes. Images on the bottom show a corresponding decreased signal intensity on apparent diffusion coefficient that is consistent with acute abnormal restricted diffusion. These findings suggest new/ongoing acute infarcts. |
On day 22, patient’s clinical status continued to deteriorate. There was no neurological improvement. Despite therapeutic anticoagulation, patient was found to have a left upper extremity arterial thrombus requiring emergent thrombectomy.
Hemoglobin remained stable but her platelets continued to drop. Heparin-induced thrombocytopenia was on the differential given the drop in platelets while the patient was on heparin. Therefore, heparin was changed to argatroban. Serotonin release assay was negative but PF4 antibody was positive.
On day 35, patient’s clinical status continued to deteriorate further with ongoing systemic clotting. She now had bilateral internal jugular vein clots as well as cyanotic extremities. After a palliative care meeting with the family, decision was made to withdraw care and the patient expired. An autopsy was performed given the complexity of the case. Autopsy findings were the following: ischemic necrosis of most organs including the liver, lungs, and adrenal glands; cardiomegaly with atrial and ventricular thrombi, mitral vegetation with pericardial effusion; right lung hemorrhage infarct with vascular emboli; hepatomegaly with congestion, necrosis; kidneys with extensive necrosis and vascular emboli; mitral valve vegetation, 10 cm in size, had lots of necrosis and the mitral valve tissue showed focal necrosis without the presence of bacterial or fungal organisms; myocardium was normal without lymphocytic infiltrates or granulomas; clusters of fibrinous thrombi with lymphoid myeloid cells adjacent to the myocardial tissue possibly related to extensive vegetations. These findings were consistent with non-bacterial thrombotic endocarditis (Figs. 4-8).
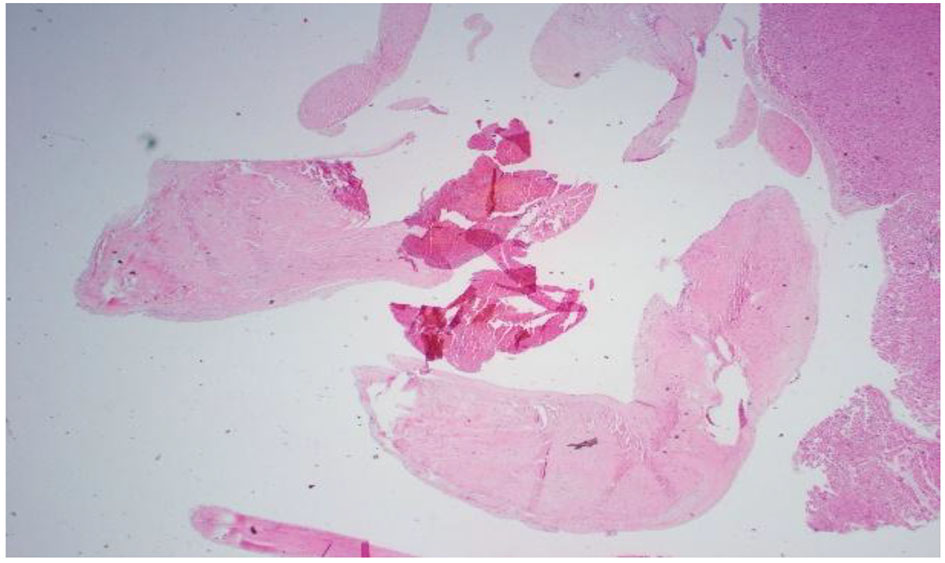 Click for large image | Figure 4. Pathology slide of mitral valve vegetation. Lots of necrosis: 10 cm circumference vegetation. Mitral valve tissue shows focal necrosis. No bacterial or fungal organisms were present. |
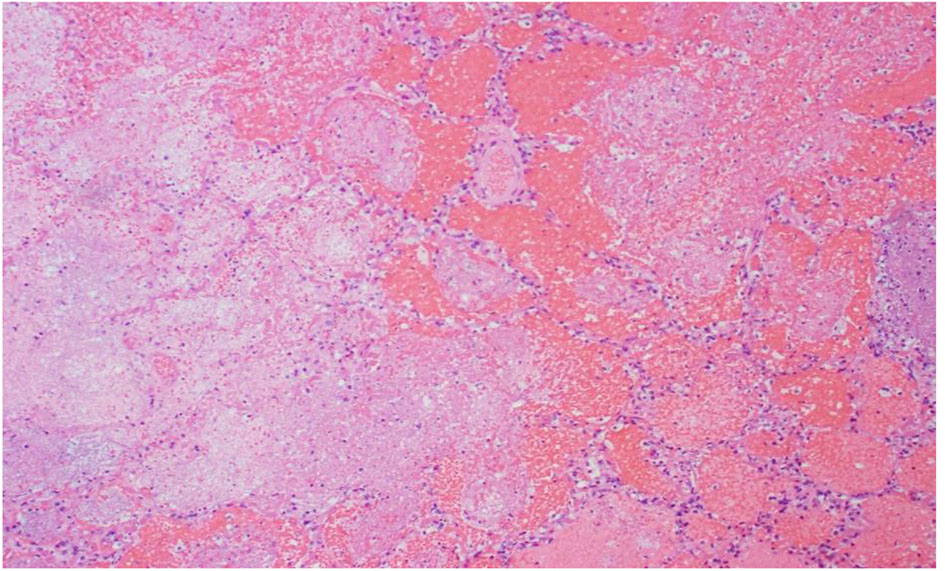 Click for large image | Figure 5. R lung, high power: emboli and large necrotic infarcted tissue. |
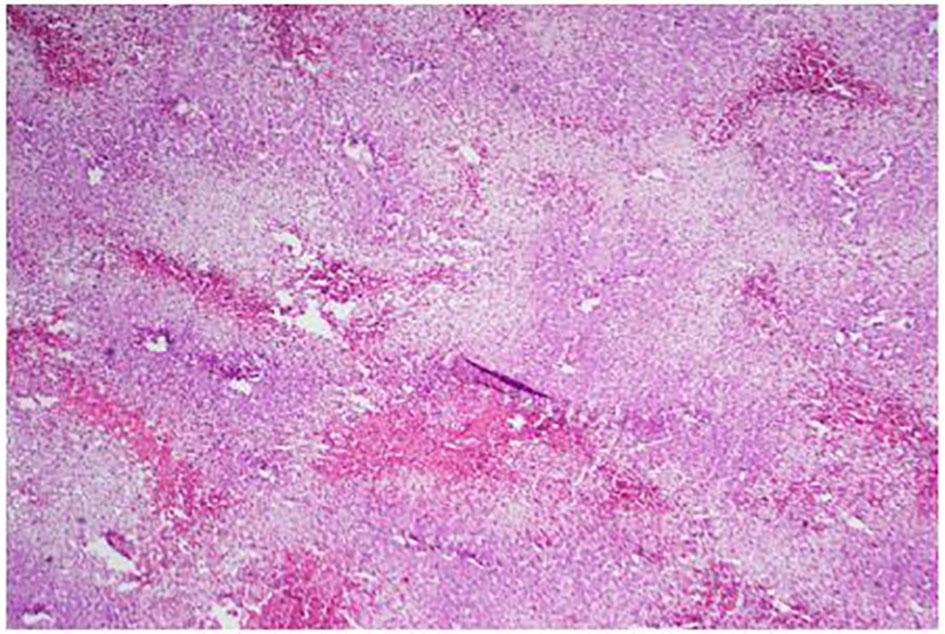 Click for large image | Figure 6. Low power of the liver: lots of steatosis and congestion, necrosis. |
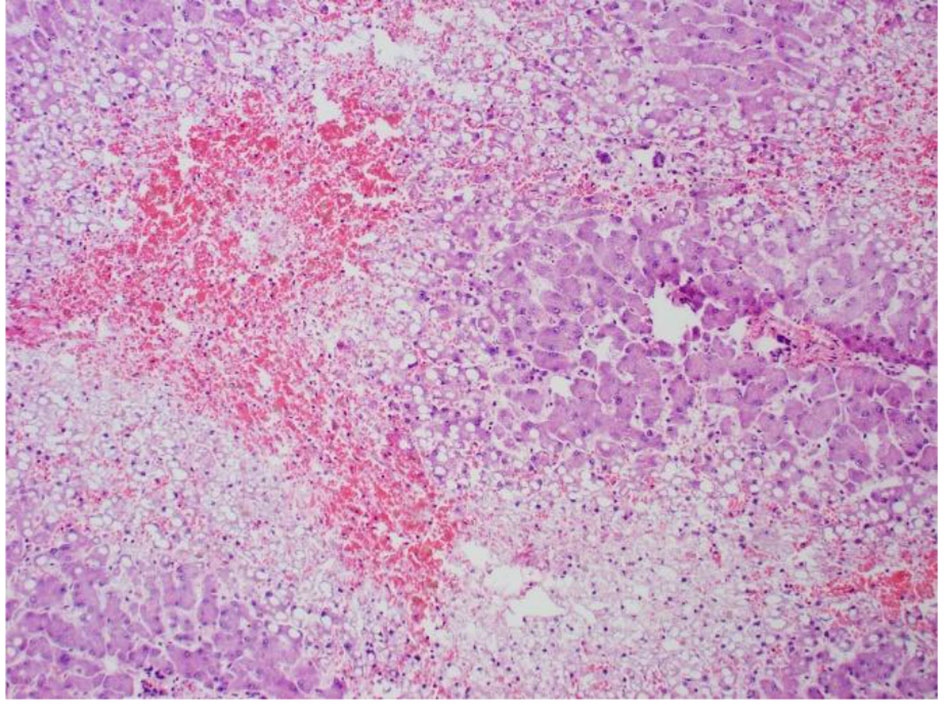 Click for large image | Figure 7. High power pathology slide of the liver showing lots of steatosis, congestion, and necrosis. |
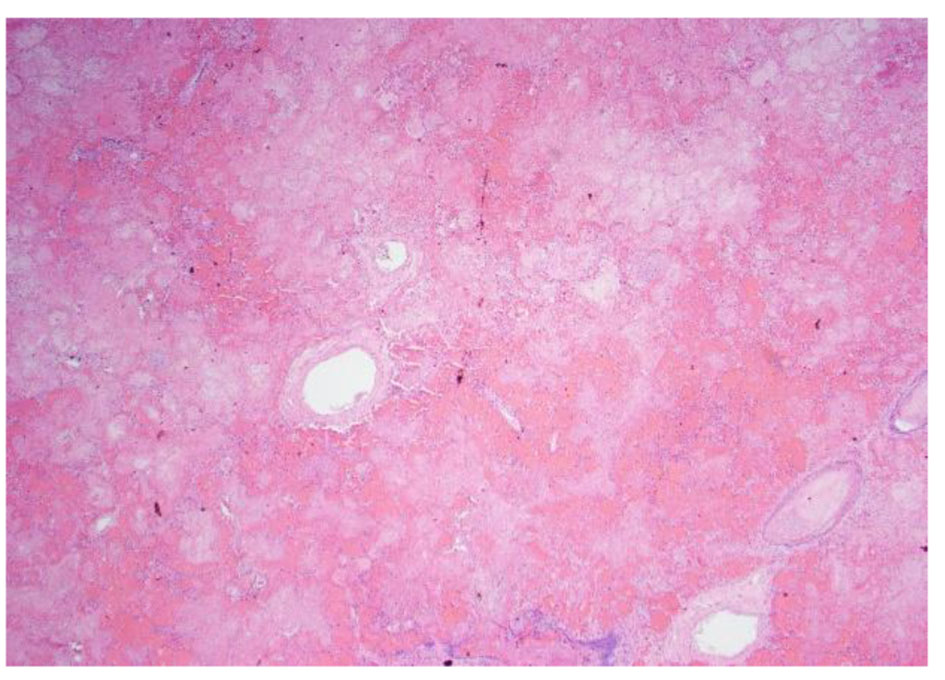 Click for large image | Figure 8. Low power pathology slide of the lung showing emboli and necrotic tissue. |
| Discussion | ▴Top |
APS is a systemic autoimmune disease that results in a prothrombotic state. Thrombosis can occur in the arterial or venous circulation [4]. It can also be associated with loss of pregnancy. Characteristic laboratory abnormalities in APS include elevated levels of antibodies against the membrane phospholipids and/or their associated plasma proteins [5]. Laboratory criteria for diagnosis of APS involve the presence of one or more antiphospholipid antibodies measured on two or more occasions at least 12 weeks apart. Antiphospholipid antibodies include anticardiolipin antibodies, lupus anticoagulant, and anti-beta2-glycoprotein. These laboratory findings along with clinical criteria of vascular thrombosis or pregnancy mortality are used for the diagnosis of APS [6]. APS can occur as a primary disorder or secondary to an underlying disease such as SLE or other systemic autoimmune diseases. Our patient’s laboratory workup was negative for any associated autoimmune diseases.
CAPS is the most severe manifestation of APS and tends to occur in 1% of patients with APS. It is characterized by the presence of thrombosis in multiple organs. It is defined as thrombosis of at least three different organs systems over a short period of time with histopathologic evidence of ischemia, vessel occlusions and high titers of antiphospholipid antibodies [7]. Diagnosis of CAPS requires the following manifestations: involvement of three organs or systems, development of symptoms within less than 1 week, confirmation by histopathology of small vessel occlusion in at least one organ tissue, and the presence of antiphospholipid antibodies. Definite CAPS requires the presence of all of these [8]. The main autopsy finding was microthrombosis in most patients. Our patient met the criteria for CAPS given the autopsy findings.
In our case, it was important to differentiate CAPS from other fatal but similar presentation. The differentiation between hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP) and CAPS is sometimes difficult. While thrombocytopenia and schistocytes are marked in HUS/TTP, they are usually absent in CAPS. Activated partial thromboplastin time is usually normal in HUS/TTS, but it may be elevated in CAPS in the presence of lupus anticoagulant [9].
CAPS is usually triggered by a precipitating factor, most likely an infection in younger patients, and malignancy in older patients. Majority of patients with CAPS have an underlying autoimmune disease. Any organ can be affected. However, the most common organs affected are kidneys, lungs, and the brain. On the other hand, patients with lupus tend to have more cardiac and brain involvement [10].
Finally, management of CAPS is complex because as there are numerous thrombotic events, the clinical presentation can be difficult to differentiate from that of disseminated intravascular coagulation, and the optimal treatment regimen is unknown. When CAPS is suspected, anticoagulation and steroids should be started. Treatment of precipitating factors such as sepsis should be considered. Anticoagulation, corticosteroids, plasmapheresis, and intravenous immunoglobulin (IVIG) are most commonly used in the management of these patients. Other reported management options for CAPS patients include fibrinolytics, prostacyclin, defibrotide, danazol, cyclophosphamide, and azathioprine [11]. Looking back in retrospect, we could have treated our patient with either immunosuppression and/or plasmapheresis or steroids. She was being anticoagulated without any improvement and further clotting.
Libman-Sacks endocarditis, or NBTE, was first described in 1924 by Libman and Sacks [12]. It is characterized by sterile vegetations deposited on heart valves, often favoring the left side of the heart. These vegetations consist of fibrin mixed with immune complexes and platelets [1]. Endothelial damage in the setting of a hypercoagulable state is thought to be a precipitating factor in NBTE [13]. NBTE is commonly associated with malignancy and SLE. Other inflammatory states such as antiphospholipid syndrome, sepsis, and rheumatoid arthritis can also be settings for NBTE.
Meanwhile, inflammatory changes can involve all layers of the heart in SLE patients with Libman-Sacks being the most common valvular abnormality. Valvular abnormalities have been seen in up to 35% of patients with SLE and up to 48% in lupus patients with APS [14].
Libman-Sacks endocarditis is often times diagnosed on autopsy as symptoms present later in the disease progression. High clinical suspicion is necessary for diagnosis. Advancements in echocardiography have allowed for earlier diagnosis of NBTE [15]. Patients with NBTE typically present with thrombotic events. NBTE can lead to significant complications such as cerebrovascular embolism and systemic thromboembolisms [16]. Patients may also present with cardiac failure from valvular dysfunction along with generalized symptoms of fatigue, fevers, night sweats, and weight loss. Our patient presented in acute heart failure and cardiogenic shock with severe valvular disease. Vegetations were seen on the TEE. Our patient also had diffuse systemic emboli as evidenced on CT of head and MRI of brain as well as autopsy findings. CAPS can be a life-threatening presentation of APS and high clinical index of suspicion is necessary to guide the diagnosis. As in our patient, diagnosis is often made from the autopsy. High clinical suspicion is required and early treatment should be instituted to potentially reverse this highly morbid condition.
Financial Disclosures
We do not have any financial disclosures nor have we received any funding.
Conflicts of Interest
There are no conflicts of interest.
Consent
Consent was obtained.
| References | ▴Top |
- Menard GE. Establishing the diagnosis of Libman-Sacks endocarditis in systemic lupus erythematosus. J Gen Intern Med. 2008;23(6):883-886.
doi pubmed - Blank M, Shani A, Goldberg I, Kopolovic J, Amigo MC, Magrini L, Shoenfeld Y. Libman-Sacks endocarditis associated with antiphospholipid syndrome and infection. Thromb Res. 2004;114(5-6):589-592.
doi pubmed - Hojnik M, George J, Ziporen L, Shoenfeld Y. Heart valve involvement (Libman-Sacks endocarditis) in the antiphospholipid syndrome. Circulation. 1996;93(8):1579-1587.
doi pubmed - Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033-1044.
doi pubmed - Garcia D, Akl EA, Carr R, Kearon C. Antiphospholipid antibodies and the risk of recurrence after a first episode of venous thromboembolism: a systematic review. Blood. 2013;122(5):817-824.
doi pubmed - Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295-306.
doi pubmed - El-Shereef RR, El-Abedin Z, Abdel Aziz R, Talat I, Saleh M, Abdel-Samia H, Sameh A, et al. Catastrophic Antiphospholipid Syndrome. Case Rep Rheumatol. 2016;2016:4161439.
doi - Erkan D, Cervera R, Asherson RA. Catastrophic antiphospholipid syndrome: where do we stand? Arthritis Rheum. 2003;48(12):3320-3327.
doi pubmed - Nayer A, Ortega LM. Catastrophic antiphospholipid syndrome: a clinical review. J Nephropathol. 2014;3(1):9-17.
pubmed - Rodriguez-Pinto I, Moitinho M, Santacreu I, Shoenfeld Y, Erkan D, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun Rev. 2016;15(12):1120-1124.
doi pubmed - Bucciarelli S, Espinosa G, Cervera R, Erkan D, Gomez-Puerta JA, Ramos-Casals M, Font J, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54(8):2568-2576.
doi pubmed - Lee JL, Naguwa SM, Cheema GS, Gershwin ME. Revisiting Libman-Sacks endocarditis: a historical review and update. Clin Rev Allergy Immunol. 2009;36(2-3):126-130.
doi pubmed - Liu J, Frishman WH. Nonbacterial Thrombotic Endocarditis: Pathogenesis, Diagnosis, and Management. Cardiol Rev. 2016;24(5):244-247.
doi pubmed - Kasar PA, Mathew M, Abraham G, Kumar RS. Occult systemic lupus erythematosus with active lupus nephritis presenting as Libman-Sacks endocarditis. Ann Pediatr Cardiol. 2012;5(1):85-88.
doi pubmed - Asopa S, Patel A, Khan OA, Sharma R, Ohri SK. Non-bacterial thrombotic endocarditis. Eur J Cardiothorac Surg. 2007;32(5):696-701.
doi pubmed - Aryana A, Esterbrooks DJ, Morris PC. Nonbacterial thrombotic endocarditis with recurrent embolic events as manifestation of ovarian neoplasm. J Gen Intern Med. 2006;21(12):C12-15.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.