Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access
Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc
Journal website http://www.cardiologyres.org
Case Report
Volume 8, Number 3, June 2017, pages 117-122
Heterozygous Familial Hypercholesterolemia With APOE Haplotype: A Prospective Harbinger of a Catastrophic Cardiovascular Event
Glenmore Lasama, c, Siddesh Shambhua, Robert Fishbergb
aDepartment of Medicine, Atlantic Health System-Overlook Medical Center, Summit, NJ 07901 USA bSection of Cardiology, Atlantic Health System-Overlook Medical Center, Summit, NJ 07901 USA cCorresponding Author: Glenmore Lasam, Department of Medicine, Overlook Medical Center, Summit, NJ 07901, USA
Manuscript submitted April 19, 2017, accepted May 22, 2017 Short title: Heterozygous FH With APOE doi: https://doi.org/10.14740/cr548w
We report a very young man with heterozygous familial hypercholesterolemia (FH) with APOE haplotype and a significant cardiac family history who underwent cardiac catheterization for intermittent episodes of exertional dyspnea and was noted to have a severe triple vessel coronary artery disease (CAD). He underwent coronary artery bypass graft (CABG) surgery which was uneventful. He was discharged on antiplatelet, beta blocker, nitrate, and statin. On routine health maintenance evaluation, he had no cardiac complaints and had been tolerating well his activities of daily living.
Familial hypercholesterolemia (FH) is a silent genetic disorder that predisposes an individual with no predilection to age to premature coronary artery disease (CAD). Early on, aggressive anticholesterol medication should be instituted. However, in cases of individuals with manifestations of heart disease that has been confirmed on angiography, coronary artery bypass graft (CABG) should be done to halt the progression of symptoms and prevent deleterious complications.
A 27-year-old male with a medical history significant for FH diagnosed approximately 10 years ago presented for further evaluation. He had been diagnosed in 2006 with a particularly virulent genetic form of heterozygous FH involving mutations in low-density lipoprotein (LDL) receptor with Gln125 variant and APOE haplotype E3/E4. He had a strong family history of CAD. He was noted to be intolerant to several statins but had recently been tolerating simvastatin. Other pertinent medical history included only seasonal allergies. Current medications being taken included simvastatin, ezetimibe, evolocumab, aspirin, alprazolam, and albuterol.
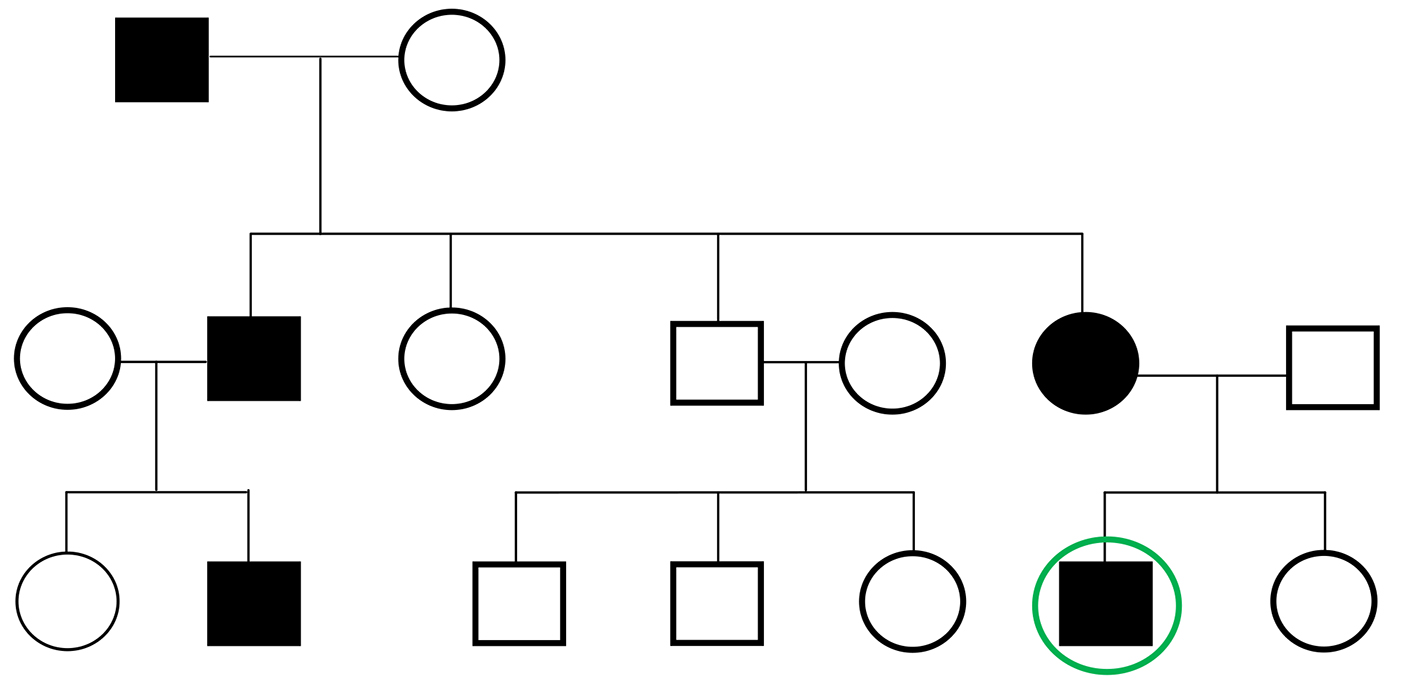
The patient had significant family history including a recently diagnosed mother with heterozygous FH, a cousin who had a sudden death at age 29 from a myocardial infarction and a grandfather and an uncle who both required CABG in their 40s (Fig. 1). Social history revealed that the patient was an ex-smoker having quit at the age of 22 and he rarely drank alcohol. Review of systems was significant for dyspnea going up stairs but with good exercise tolerance however. The patient denied any chest pain on exertion to imply possible unstable angina.
Figure 1. Pedigree of the patient depicting his grandfather and uncle who possibly have FH in the setting of heart attack in their 40s requiring CABG, his mother with recently diagnosed heterozygous FH, and a cousin who had a sudden cardiac death at age 29.
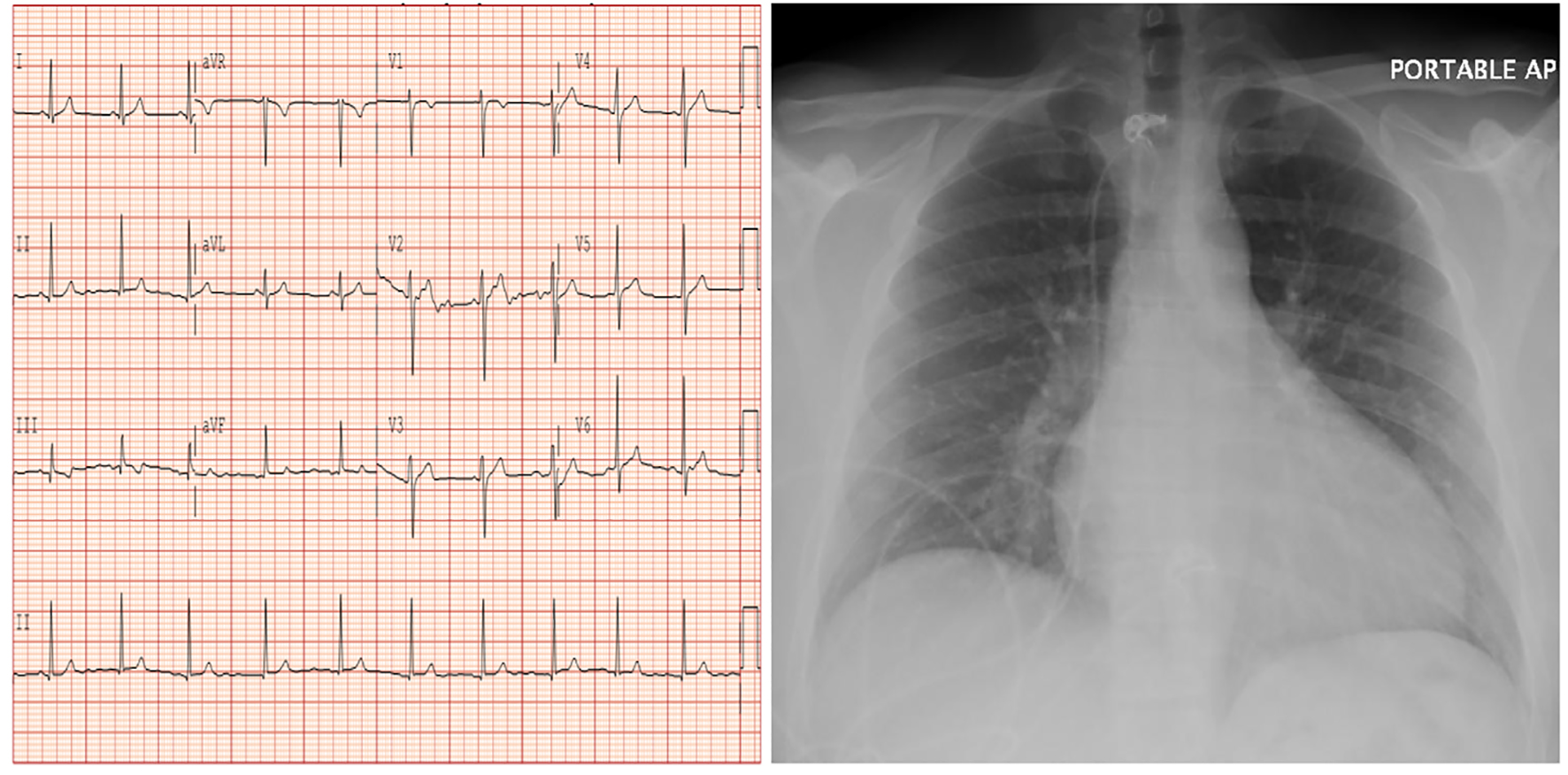
On physical examination, the patient’s heart rate was in the 60s in regular rhythm and his blood pressure was normal. He has moist oral mucosa, clear conjunctiva, and a note of a gray arc on the upper area of his cornea. There were no palpable head or neck masses and heart sounds were normal with no murmurs. Neither carotid nor abdominal bruit was appreciated. Abdomen was soft, non-tender with no masses and peripheral pulses were normal with no peripheral cyanosis or clubbing of his extremities. Neurological exam was unremarkable. Electrocardiogram revealed sinus rhythm with no ischemic changes while chest radiograph showed no acute cardiopulmonary process but had a prominent heart (Fig. 2).
Figure 2. Electrocardiogram showing sinus rhythm with no signs of ischemia (left panel) while chest radiograph (right panel) revealing prominent heart with no acute cardiopulmonary process.
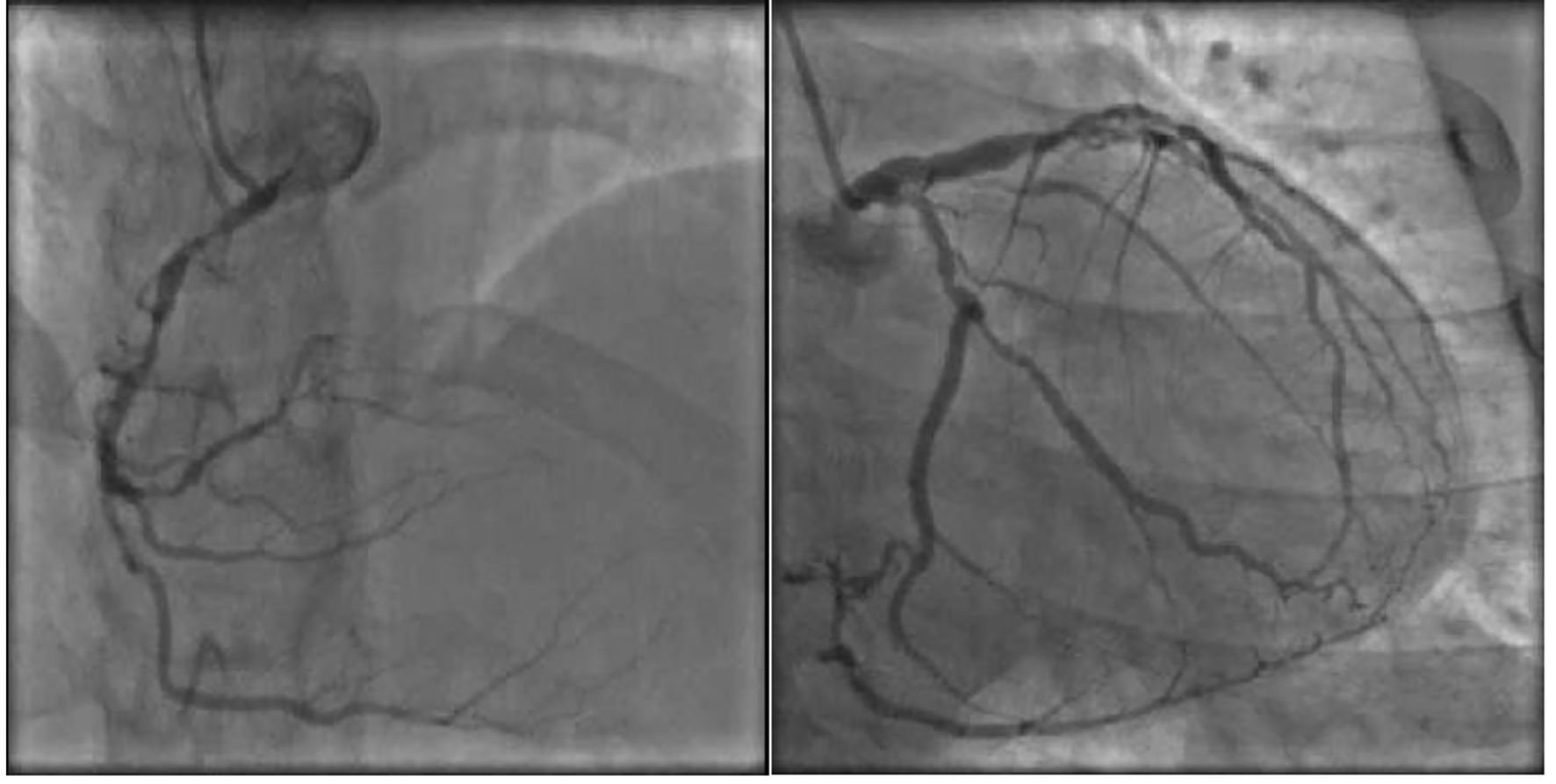
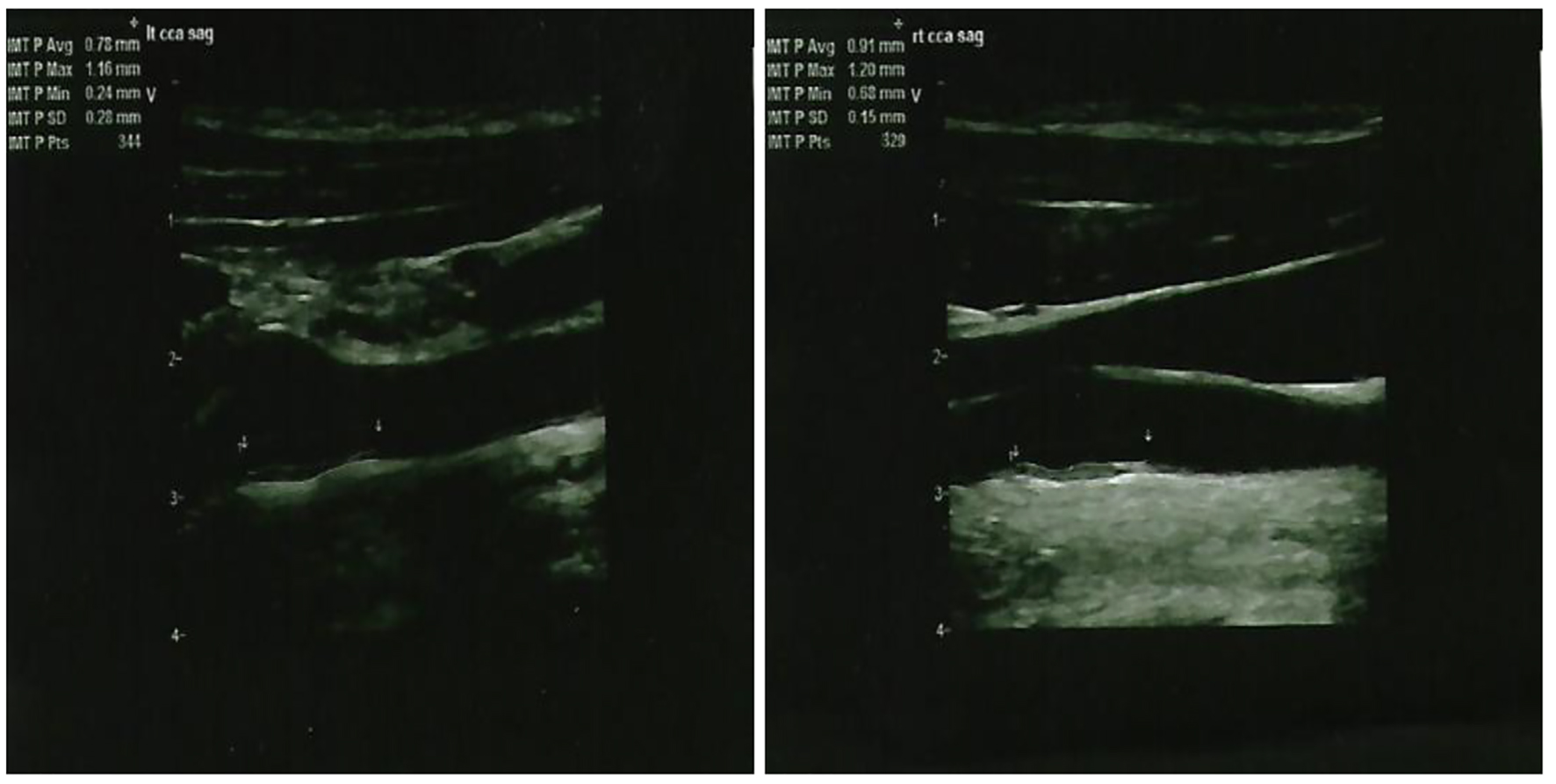
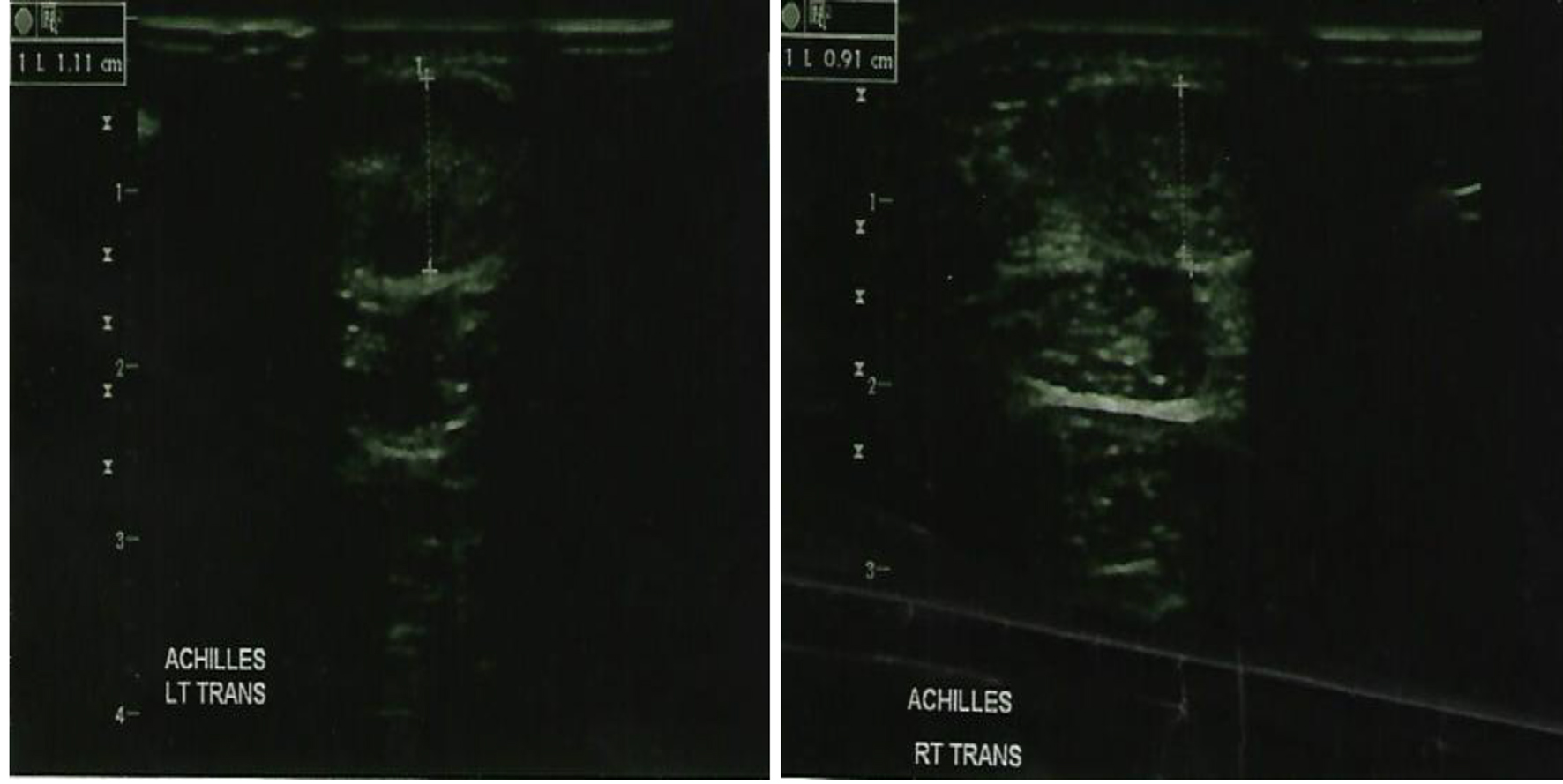
Given his strong family history of premature CAD and FH, the patient underwent a nuclear stress test which was positive for evidence of reversible ischemia. For further evaluation, the patient underwent cardiac catheterization which revealed severe triple vessel CAD with critical lesions in the proximal to mid left anterior descending (90%), first (60%) and second (90%) diagonal branches, proximal circumflex (95%) and first obtuse marginal (80%), the proximal right coronary artery (80%), and the proximal right posterolateral branch (99%) together with chronic total obstruction of the distal right coronary artery (Fig. 3). The patient did have preserved left ventricular function however. A bilateral carotid ultrasound was done which showed a left common carotid intima-media thickness (CIMT) range of 0.28 - 1.16 mm, while a right common CIMT range of 0.68 - 1.20 mm, which were both leaning towards the abnormal value of > 0.9 mm (Fig. 4). Also, an achilles tendon (AT) ultrasound was done to assess its thickness which revealed 1.11 cm on the left and 0.91 cm on the right, both above the threshold value of 0.53 cm (Fig. 5).
Figure 4. Ultrasound showing the left common carotid intima-media thickness (left panel) and right common carotid intima-media thickness (right panel).
Figure 5. Ultrasound showing the left achilles tendon thickness (left panel) and right achilles tendon thickness (right panel).
Following this due to the extensive nature of complex disease, the patient was referred for CABG. The patient subsequently went for cardiac surgery with the use of the right internal mammary artery to the left anterior descending, left internal mammary artery to the circumflex marginal artery, radial artery graft from the ascending aorta to the right posterolateral branch of the right coronary artery, saphenous vein graft from ascending aorta to the second diagonal artery and saphenous vein graft from the aorta to the right acute marginal branch of the right coronary artery. No significant valvular pathology was noted and post-procedure ejection fraction was between 55% and 60%. The patient’s postoperative recovery was otherwise unremarkable and he was discharged home on aspirin, atenolol, isosorbide, and simvastatin and emphasized regular health maintenance evaluation by his cardiologist.
FH is a genetic disorder which predisposes to premature atherosclerosis that eventually leads to early onset coronary heart disease (CHD) to include angina and myocardial infarction. It is an autosomal dominant disease due to mutations in the low-density lipoprotein receptor gene (LDLR), the apolipoprotein B-100 gene (APOB), and the newly discovered proprotein convertase subtilisin/kexin type 9 gene (PCSK9) [1]. PCSK9 is a serine protease released into the plasma from the liver which binds primarily to the LDLR in the liver cells [2]. The end result is to prevent the recycling to the cell surface of the LDLR and eventually stimulate disintegration within the hepatocyte [3]. A rare autosomal recessive type has been elucidated that involves an adaptor protein defect for the LDLR [4]. All these mechanisms culminate to a rise in plasma LDL-cholesterol (LDL-C) because of diminished removal of LDL molecules in the circulation. Alongside, the macrophage receptor scavenges and facilitates the ingestion of oxidized LDL that generates the formation of foam cells and accumulation of lipids in macrophages that deposit beneath the endothelium which predisposes to atheroma formation [5].
FH has been estimated among white population to be 1:1,000,000 in homozygous, while 1:250 - 500 in heterozygous [1, 6]. In most countries, less than 1% has been diagnosed, while 6-79% have been identified in European nations though it is underdiagnosed and undertreated in the general population [7].
FH diagnosis is based on five criteria, namely family history, premature CHD, xanthomas and corneal arcus on physical examination, consistently very high LDL-C level, and/or confirmed genetic mutation [8]. AT sonography increases the clinical diagnosis of AT xanthomas from 43% to 68% with thickness threshold for the diagnosis of FH of 5.3 and 5.7 mm in men less than and more than 45 years, and 4.8 and 4.9 mm in women less than and more than 50 years, respectively [9]. CIMT measurement combined with plaque assessment is an excellent measure of cardiovascular risk but has conflicting recommendations regarding its utilization. CIMT has been recommended in 2010 by the American Heart Association/American College of Cardiology (AHA/ACC) for cardiovascular risk assessment in intermediate risk asymptomatic adults (class IIa) [10], however, refuted in 2013 regarding its use in clinical practice for risk assessment for a first atherosclerotic cardiovascular disease event [11].
The severity of hypercholesterolemia in FH homozygotes usually culminates into critical atherosclerosis causing cardiovascular disease during childhood and adolescence [12]. Calcification of the coronary artery which has been unusually determined prior to fourth decade was established in a considerable percentage in heterozygotes at 11 - 23 years of age [13]. Aortic valve stenosis has often been described in homozygous FH [14], though it is infrequent in heterozygous [15].
Our patient is unique because of his heterozygous FH involving mutations in LDLR with Gln125 variant and APOE haplotype E3/E4 which could have predisposed him early to CAD. The prevalence of ischemic heart disease was significantly greater in FH patients with apoE4 (73.3%) compared to those without apoE4 (31.4%) [16]. The odds ratio for CHD risk with the *ε4 compared with the *ε3 allele was 1.38 for men, while 1.82 for women [17]. A 40% increased risk for CHD mortality has been estimated for *ε4 carriers compared with *ε3*ε3 genotype or *ε2 carriers [18]. The biochemical mechanism has been associated to an alteration in lipoprotein metabolism by apoE4 with subsequent increase in LDL concentration which links apoE4’s predisposition to CAD [19].
CABG in young FH patient is challenging and very seldom in the first and second decade though feasible and has good outcomes. Two cases have been reported of a 13- and a 14-year-old boy with FH who underwent triple-vessel CABG utilizing bilateral internal thoracic artery and saphenous vein grafting devoid of unfavorable events [20, 21]. The role of CABG in FH patients with documented CAD has been elucidated in literature.
Cardiac event-free rate in a 10-year period after CABG was 58% and results were as good in FH as in non-FH patients using arterial grafts and anticholesterol therapy [22]. Appraisal of coronary bypass operations in FH had a cardiac free event of 60% at 5 and 10 years for homozygotes compared to 87% and 73% for heterozygotes which has been comparable with patients without FH who were randomly age-matched that underwent CABG during the same time frame [23].
Albeit of severe coronary atherosclerosis, patients with FH showed good long-term outcome after CABG with no cardiac death during a mean follow-up period of 52 months and with an actuarial survival rate of 95% at 5 years and 89% at 12 years after procedure [24].
Additionally, all FH patients should initiate lifestyle modification and receive drug therapy to diminish cardiovascular risk, taking into account smoking, family background, intensity of LDL-C elevation, and the emergence of other cardiovascular comorbidities which should dictate the extent of treatment [25]. The addition of the PCSK9 inhibitor evolocumab to statin therapy decreased LDL-C levels by 59% from baseline levels and remarkably reduced by 15-20% the risk of cardiovascular events [26]. A novel antisense oligonucleotide, mipomersen, is a favorable therapeutic alternative to lower LDL-C, apoB, and total cholesterol levels in patients with uncontrolled FH whose LDL-C targets are not sufficiently met by maximal medical treatment [27]. Also, lipoprotein apheresis remains an indication for homozygous FH and in patients with progressive coronary vascular disease secondary to heterozygous FH refractory to statins or to an elevated levels of lipoprotein(a) [28].
Conclusion
Close surveillance is imperative for FH patients to evaluate their response to medications, address its associated side effects, and institute aggressive surgical intervention if evidence of a significant CAD had been established.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160(5):407-420. doipubmed
Rosenson RS, de Ferranti SD, Durrington P. Inherited disorders of LDL-cholesterol metabolism. UpToDate, Waltham, MA. (Accessed on January 10, 2017).
Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(Suppl):S172-177. doipubmed
Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292(5520):1394-1398. doipubmed
Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320(14):915-924. pubmed
Goldstein JK, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds), The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. p. 2863-2913
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-3490a. doipubmed
Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173(1):55-68. doipubmed
Junyent M, Gilabert R, Zambon D, Nunez I, Vela M, Civeira F, Pocovi M, et al. The use of Achilles tendon sonography to distinguish familial hypercholesterolemia from other genetic dyslipidemias. Arterioscler Thromb Vasc Biol. 2005;25(10):2203-2208. doipubmed
Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, Foster E, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2010;56(25):e50-103. doipubmed
Goff DC, Jr., Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Sr., Gibbons R, Greenland P, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2935-2959. doipubmed
Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S1-8. doipubmed
Gidding SS, Bookstein LC, Chomka EV. Usefulness of electron beam tomography in adolescents and young adults with heterozygous familial hypercholesterolemia. Circulation. 1998;98(23):2580-2583. doipubmed
Tsuchida M, Kawashiri MA, Tada H, Takata M, Nohara A, Ino H, Inazu A, et al. Marked aortic valve stenosis progression after receiving long-term aggressive cholesterol-lowering therapy using low-density lipoprotein apheresis in a patient with familial hypercholesterolemia. Circ J. 2009;73(5):963-966. doipubmed
Rallidis L, Naoumova RP, Thompson GR, Nihoyannopoulos P. Extent and severity of atherosclerotic involvement of the aortic valve and root in familial hypercholesterolaemia. Heart. 1998;80(6):583-590. doipubmed
Eto M, Watanabe K, Chonan N, Ishii K. Familial hypercholesterolemia and apolipoprotein E4. Atherosclerosis. 1988;72(2-3):123-128. doi
Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155(6):487-495. doipubmed
Stengard JH, Weiss KM, Sing CF. An ecological study of association between coronary heart disease mortality rates in men and the relative frequencies of common allelic variations in the gene coding for apolipoprotein E. Hum Genet. 1998;103(2):234-241. doipubmed
Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8(1):1-21. doipubmed
Shirish SB, Kamath SG, Sunil SCV, Chidanand B, Nitin K. Triple Vessel Coronary Artery Bypass Grafting in a 14-year-old Child with Familial Hypercholesterolemia-A Rare Case Report. Open J Cardiovasc Surg. 2009;2:11-13.
Tuncer E, Basaran M, Cine N, Tuzun B, Ceyran H. Coronary revascularization in a 13 year old patient with homozygous familial hypercholesterolemia. TUrk Gogus Kalp Damar Cerrahisi Dergisi. 2014;22(1):168-170. doi
Tedoriya T, Kawasuji M, Sakakibara N, Ueyama K, Takemura H, Watanabe Y. Coronary artery bypass surgery in patients with familial hypercholesterolemia. Nihon Kyobu Geka Gakkai Zasshi. 1992;40(7):1095-1099. pubmed
Takahashi T, Nakano S, Shimazaki Y, Kaneko M, Hirata N, Nakamura T, Matsuzawa Y, et al. Long-term appraisal of coronary bypass operations in familial hypercholesterolemia. Ann Thorac Surg. 1993;56(3):499-505. doi
Kawasuji M, Sakakibara N, Takemura H, Matsumoto Y, Mabuchi H, Watanabe Y. Coronary artery bypass grafting in familial hypercholesterolemia. J Thorac Cardiovasc Surg. 1995;109(2):364-369. doi
Robinson JG, Goldberg AC. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S18-29. doipubmed
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doipubmed
Agarwala A, Jones P, Nambi V. The role of antisense oligonucleotide therapy in patients with familial hypercholesterolemia: risks, benefits, and management recommendations. Curr Atheroscler Rep. 2015;17(1):467. doipubmed
Walji S, Neuwirth C, Thompson GR. Lipoprotein Apheresis for the Treatment of Familial Hypercholesterolemia. Clin Lipidology. 2013;8(5):573-586. doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.